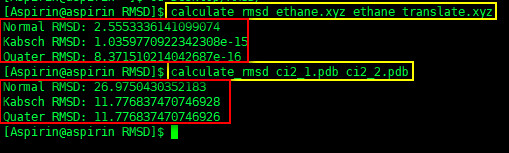
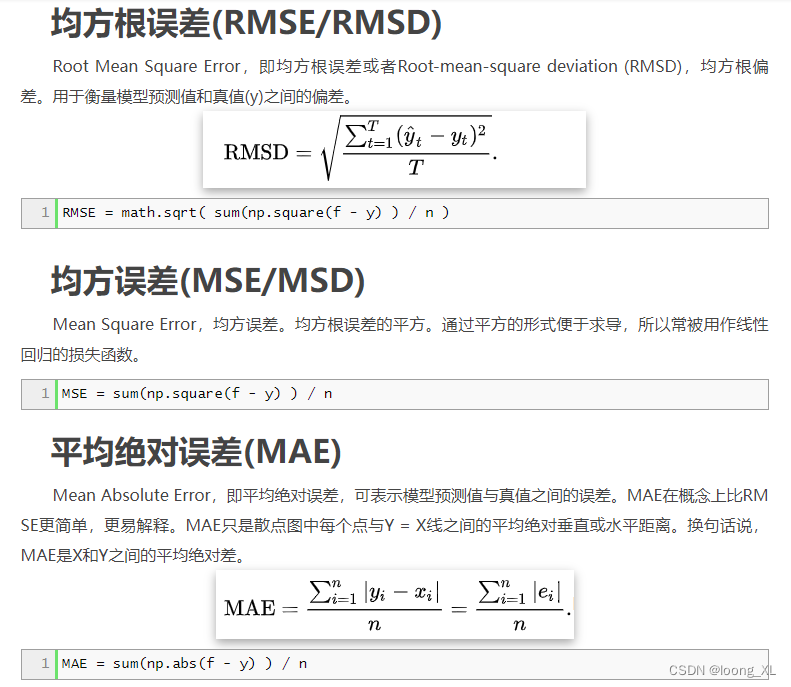
今天VS2017造成电脑死机,重启电脑后打开VS进行调试发现程序启动失败,原因是读取Settings配置信息时产生“配置系统未能初始化”异常,但是App.config文件并未修改,所以网上的一些解决办法在这里并不适用了,由于C#的配置信息会存储与用户目录中,则查找用户\AppData\Local目录下程序对应配置文件夹对应版本中的user.config文件,发现已变为非文本格式,将该文件删除后再运行程序则不再报此异常。
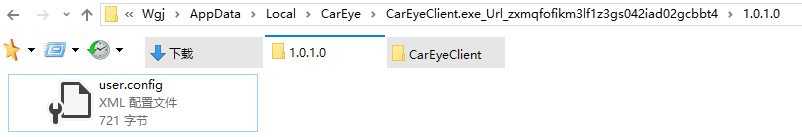
文件所在目录格式如下图:
今天VS2017造成电脑死机,重启电脑后打开VS进行调试发现程序启动失败,原因是读取Settings配置信息时产生“配置系统未能初始化”异常,但是App.config文件并未修改,所以网上的一些解决办法在这里并不适用了,由于C#的配置信息会存储与用户目录中,则查找用户\AppData\Local目录下程序对应配置文件夹对应版本中的user.config文件,发现已变为非文本格式,将该文件删除后再运行程序则不再报此异常。
文件所在目录格式如下图: