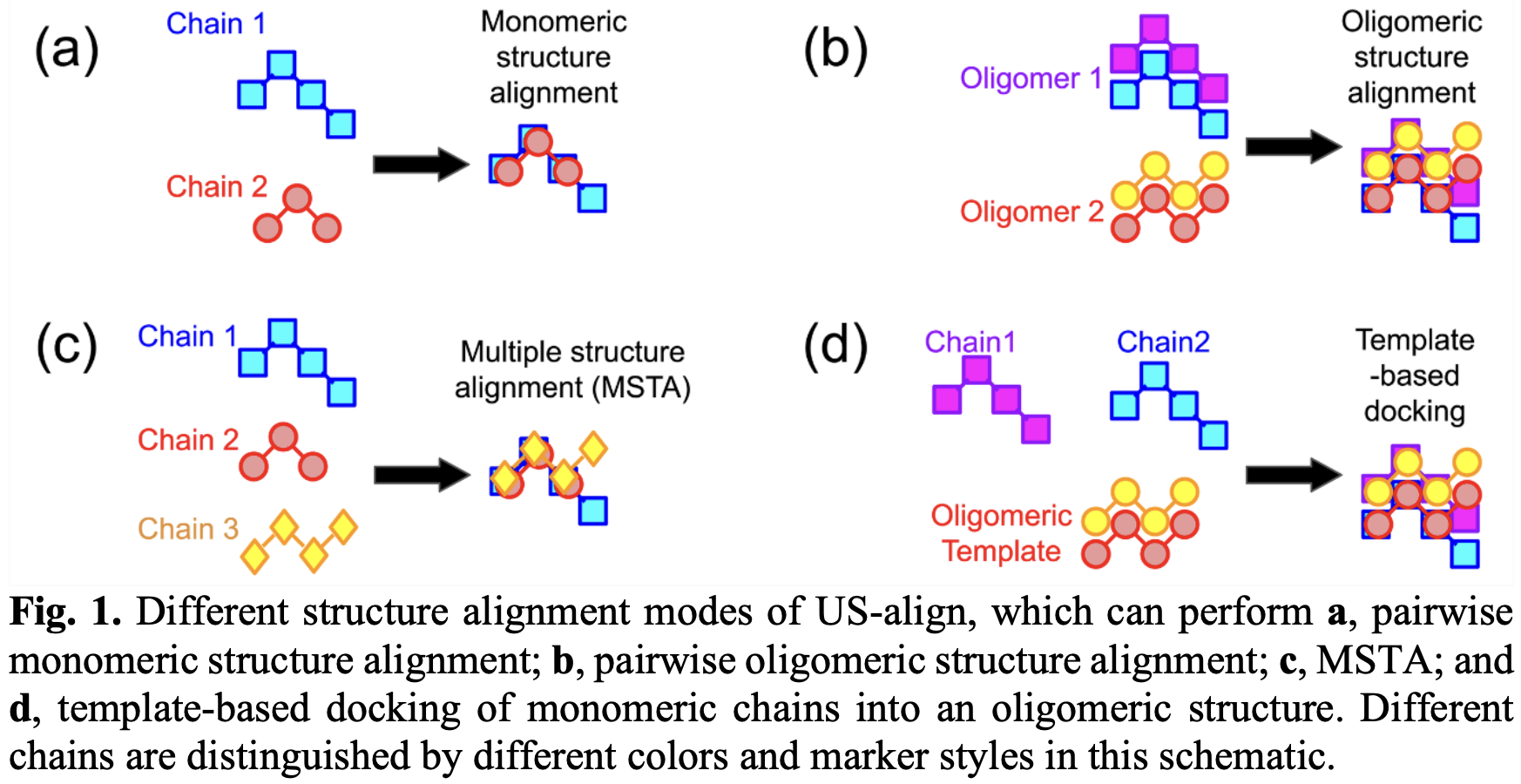
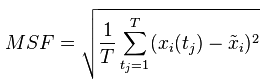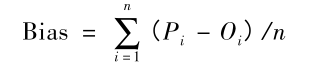Nature子刊 Machine Intelligence发布了八月份最新接收论文,共4 篇。一篇是清华生命学院龚海鹏和澳大利亚格里菲斯大学周耀旗等人用神经网络进行蛋白质结构预测方面的工作。
DeepMind提出的AlphaFold证明了深度学习能大幅度提高蛋白质结构预测准确度。2019年8月,Nature Machine Intelligence提出的DeepFragLib,有望将该方向推进到新的阶段。
1. 简介
预测蛋白质的未知天然结构的典型方法是组装从已知结构提取的氨基酸残基(片段)。提取的片段质量用于构建蛋白质特异性片段文库,并可确定采样近原始构象的成功与否。本文展示了如何使用深层上下文学习技术构建高质量的片段库。本文的算法称为DeepFragLib,采用双向长短期记忆递归神经网络,通过知识蒸馏进行初始片段分类,然后是聚合残差变换网络和循环扩张卷积用于检测近原生片段。与现有方法相比,DeepFragLib将近原生片段的位置平均比例提高了12.2%,当与Rosetta集成时,72.0%的自由建模域靶标产生更好的近原生结构。DeepFragLib完全并行化,可与结构预测程序一起使用。
活生物体依赖于不同的蛋白质在每个生物过程中执行单独的功能。通过蛋白质折叠成不同结构的能力实现了多种功能,由其氨基酸序列所决定。实验确定数亿种已知蛋白质结构的成本令人望而却步,故有必要直接从它们的序列计算预测蛋白质结构。从头算蛋白质结构预测是通过集中搜索蛋白质构象空间来定位天然结构的方法。如何有效地探索蛋白质构象空间的巨大自由度是计算结构生物学中最具挑战性的问题之一。解决该问题的一种方法是片段组装,其通过从已知结构提取的模板片段的构象来加速局部结构采样。由已知结构构建的片段构象空间认为几乎完整,因为可以使用4至16个残基的非同源片段数据库成功重建(使用正确的拓扑结构)超过一千种非冗余蛋白的集合。尽管接触辅助蛋白质折叠已经取得了进展,但片段组装仍然是从头算蛋白质结构预测中最常用的方法。
基于片段组装的模拟中,首先构建片段文库以促进构象采样。该文库在纯粹基于序列信息鉴定的靶蛋白的每个位置列出了合格的模板片段。该片段文库的质量是后续蛋白质折叠模拟的效率和准确性的基础。片段文库中近原生片段的比例(精确度)和至少一个近原生片段所覆盖的蛋白质位置比例(覆盖率)是评估最常用的指标片段库的质量。然而,对于在不同位置包含不同数量片段的片段库,“精确度”变得有偏差,特别是当在有限数量的位置富集许多片段时。为了解决这个问题,本文提出了一个称为“位置平均精度”的度量,它计算每个位置的近原始片段的比例,然后平均所有位置。这三个指标的组合可以提供更全面的质量评估。此外,基于片段组装的模拟通常在每个位置需要足够数量的片段,以便可以定位和组装不同位置的互补和补偿片段,以在蛋白质折叠模拟中进行有效的构象搜索。
最近,深度学习技术已经使得蛋白质结构预测取得实质性进展。例如,Raptor-X采用残余卷积网络来改进对蛋白质结构预测(CASP)竞争技术的关键评估中的二维接触图预测;SPOT-1D和SPOT-Contact利用一系列循环和卷积网络来提高二级结构、骨干角和残基接触的预测精度;AlphaFold通过设计用于片段生成、角度和接触预测以及具有几何约束的结构建模的各种神经网络来挑战CASP13竞争中的蛋白质结构预测领域。本项工作中,研究团队基于长短期记忆(LSTM)网络和聚合残差转换网络ResNeXt,开发了一种算法,以进一步提高片段库的质量。作为双向回归神经网络(BRNN)的变体,双向LSTM(Bi-LSTM)网络已被证明对蛋白质结构预测因子有用,因为氨基酸序列可以作为时间步骤处理。此外,ResNeXt架构基于ResNet但具有更少的超参数,通过在重复构建块中聚合具有相同拓扑的一组变换来提高许多常见任务的准确性,以便于在超深度神经网络中提取高级特征。DeepFragLib利用这些尖端技术构建包含多个分类模型和回归模型的分层体系结构。具体来说,在分类模型中采用Bi-LSTM层来处理序列信息,然后是完全连接的层和输出节点,以区分近本地和诱饵碎片。这些模型通过知识蒸馏进一步压缩和加速。然后将来自Bi-LSTM层的激活和分类模型的最终输出以及预测的残基-残基接触图馈送到一个基于ResNeXt的回归模型中以直接预测RMSD值。评估CASP11-13竞赛的独立测试蛋白质组中的所有自由建模靶标时,DeepFragLib提高了片段库的质量,超过了其他最先进的算法,包括NNMake,LRFragLib和Flib-Coevo。此外,当与Rosetta集成用于测试构象采样时,DeepFragLib在采样高质量蛋白质结构模型时优于其他算法。DeepFragLib完全并行化,只需20-30分钟即可构建100个残基靶序列的片段库。
overall flowchart of DeepFragLib
2. 方法
2.1 数据集
为了开发CLA模型,首先从PISCES中剔除956个蛋白链来创建一个小的高分辨率片段数据集,分辨率<1.5Å,R值<0.15,成对同一性<20%。将这些蛋白质链分成7-15个残基的连续片段。使用DSSP计算每个链的二级结构和溶剂可及的表面积。骨架二面角(φ和ψ)、基于Cα原子的角度(θ和τ)和残基接触信息分别从它们的PDB结构计算。HR956组中总共190万个7至15个残基的片段产生大约200亿对与训练CLA模型的数据长度相同的片段。HR956集中的每个片段还在DeepFragLib的推理阶段中充当片段库的候选模板。
2.2 RMSD和dRMS的标准化
与RMSD类似,dRMS测量原子-原子距离矩阵的差异,用于评估片段之间的结构相似性。dRMS足以比较高度灵活的区域和计算效率,因为它避免了耗时的结构叠加。然而,RMSD和dRMS都取决于氨基酸残基的数量,这阻碍了它们对具有不同长度的片段的值的比较。因此,本研究优化了两个公式,将不同长度(7-15个残基)的片段的RMSD和dRMS标准化为10个残基片段的标准值,遵循类似于Carugo和Pongor工作的方案。对于每个片段长度,随机选择1亿个片段对并计算它们的RMSD值。RMSD分布的向右移位显示RMSD值对片段长度的依赖性。然后根据片段长度绘制各种分布的百分点,并发现明显的线性依赖性。将RMSD百分位数值除以RMSD参考值之后,所有曲线都折叠成单个曲线。使用线性和对数函数拟合曲线,发现线性函数达到最高相关系数(> 0.999)。
2.3 分类模块
第一个模块中使用堆叠的Bi-LSTM网络来构建多个繁琐的分类模型,每个模型针对一个特定的片段长度进行优化,其中蛋白质片段的一维特征作为输入。具体而言,氨基酸对之间的相似性得分来自BLOSUM62矩阵,20个氨基酸的10维物理化学特征来自Kidera等人的研究,三级通过PSIPRED预测二级结构概率,而SPIDER3预测溶剂可及的表面积、主链二面角(φ和ψ)以及基于Cα原子的角度(θ和τ)。所有上述特征被展平为43N维输入特征向量,其中N代表片段长度。采用了相同的程序来标记之前工作中使用的样本。对于每个特定片段,将所选片段与相同长度的所有其他片段(同时需要dRMS <1)之间的最低dRMS标记为20对,作为阳性样品,仔细去除片段对索引中的冗余。将所有剩余的片段对标记为阴性样品。随机选择80%的样本作为五倍交叉验证的训练集,剩余的20%的样本作为独立的测试集。
繁琐的分类模型由三个Bi-LSTM层组成,每个方向有64个节点,接着是三个额外的完全连接的隐藏层,其中有128个节点(用于处理Bi-LSTM层的最后一步输出)。Bi-LSTM层使用双曲正切(tanh)激活,而完全连接的层采用整流线性单元(ReLU)激活。该模型具有单个输出节点,具有S形激活函数,以将置信度值归一化为[0,1]。为了降低过度拟合的风险,我们在训练期间对完全连接的层施加了50%的比率,并且对所有可训练参数的L2权重正则化应用。使用交叉熵作为损失函数训练模型,其由Adam优化器优化,初始学习率为7.5e-4。
2.4 知识蒸馏
繁琐的分类模型在预测步骤中计算量过大:对于100个残基的靶标蛋白(190万×90),接近1.8亿次。因此采用知识蒸馏来训练模型压缩和加速的小模型。蒸馏模型包含一维卷积层,其具有43个内核大小为3的滤波器,接着是每个方向具有64个节点的Bi-LSTM层和具有32个节点的完全连接层。输出层与繁琐的分类模型相同。研究人员还在完全连接层上应用了50%的dropout rate,并在所有可训练参数上应用L2正则化以训练蒸馏模型。
2.5 回归模块
第二个模块中,采用了基于ResNeXt架构的超深度回归模型来直接预测候选模板片段和原生片段之间的RMSD值。对于SCOP350集合中的每个蛋白质,选择每个片段长度的每个位置由CLA模型输出的最高置信度值的前1,000个片段,其总共组成数据集含有4.3亿个样本。随机选择70%进行训练和交叉验证,其余30%作为独立测试集。训练样本进一步分为训练集和验证集,比例为4:1。改进了预测的片段残基-残基接触基质,将它们展平成载体并计算一对片段的接触载体之间的RMSD。考虑到不同片段长度的单个CLA模型在Bi-LSTM层中具有不同数量的状态(从7到15),从Bi-LSTM层统一提取所有CLA模型的最后七个时间步长的输出。CLA模型的输出,接触矢量的距离和片段长度与Bi-LSTM输出一起作为REG模型的输入进行广播。因此,输入特征具有与包含七个时间步长的Bi-LSTM输出相同的时间维度。在推断过程中从每个位置的CLA模型中选择了前5,000个片段,以通过REG模型预测RMSD。
考虑到输入特征被分解为七个时间步长,我们设计了一个循环扩张卷积,以将感知域扩展到单个层中的整个序列。我们不是填充零值,而是填充第一步开头的最后几个步骤的特征值,然后在最后一步结束时填充前几个步骤。因此,七个步骤的特征形成一个循环,并且这些特征循环地重复。对于指定的步骤,具有一,二和三的膨胀率的卷积运算可以共同将接收场扩展到所有七个步骤。在瓶颈层之后,我们设计了12个循环扩张的卷积层,每个扩张速率并联256个滤波器。因此,REG模型的一个构建块包括36个并行路径。这些构建块重复40次,然后是输出层。REG模型中也采用批量归一化和L2正则化。我们使用Adam优化器,学习率为5e-5进行模型训练。在这项工作中,所有深度神经网络都使用TensorFlow框架进行编码,并在16个1080Ti图形处理单元上进行训练。
2.6 片段选择模块
此模块中选择策略包含三个阶段,以在每个序列位置获得候选片段以构建片段库。三阶段选择策略在CASP10训练集上进行了优化。阶段1中使用一组针对不同片段长度的定制阈值来基于REG模型输出的预测RMSD值来提取候选片段。这些片段按预测的RMSD的升序排列,只保留最高的片段以确保每个位置选择的候选者数量(记录为“NC”)从未超过200.如果NC在某个位置小于50,这是尤其是当在截止阈值内预测的片段不足时,我们引入了两个额外的富集阶段。在阶段2中,以预测的RMSD的升序提取具有预测的RMSD小于相应片段长度的较低定制阈值的片段,并且一旦NC达到50,则浓缩停止。如果在阶段2,阶段3之后NC仍然小于50开始时,所有未选择的7个残基片段按照CLA模型的输出值的降序排列,并且招募顶部片段直到NC达到50.在该选择策略之后,每个位置的候选片段的数量将落在50和50之间。200.值得注意的是,第1阶段和第2阶段采用的定制RMSD阈值是根据REG模型计算的。对于每个片段长度,将阶段1阈值设定为RMSD截止值,使得用比该截止值更低的RMSD预测的90%的片段是RMSD≤2.0的近天然片段。阶段2阈值的计算方法类似,但使用较低百分比的85%。所有阈值最终控制在≤2.0Å。
2.7 片段库的质量评估
本研究采用了广泛使用的度量精度和覆盖率来评估片段库的质量。精确度定义为整个片段文库中近天然片段的分数,覆盖度定义为目标蛋白的序列位置的分数,其由至少一个来自片段文库的近天然片段覆盖,其中近原生片段是具有小于某个阈值的原始结构的RMSD值的片段。使用了一系列阈值,范围从0.1到2.0,步长为0.1,可以产生精度和覆盖范围的曲线。此外,引入了额外的度量,即位置平均精度,其平均所有序列位置上的近原始片段的分数。该度量可以消除由于在不同位置具有显着不同数量的片段而引起的人为影响。
2.8 使用Rosetta进行构象采样和模型生成
使用Rosetta v3.8 C ++源代码来评估片段文库在从头算蛋白质结构预测中的有用性。Rosetta使用蒙特卡罗模拟迭代地利用短(3-mer)和长(9-mer)片段进行片段组装。每个步骤中,随机选择的靶蛋白区段的主链扭转角将被来自片段文库的随机选择的片段替换,并且在Metropolis-Hastings算法之后将接受或拒绝所提出的构象变化。最初的Rosetta只允许长片段的固定长度。因此,仅修改标准Rosetta AbinitioRelax应用程序以接受可变长度的长片段;将DeepFragLib、NNMake和FlibCoevo构建的片段库转换为Rosetta片段格式,并将它们直接送入AbinitioRelax,分别替换标准的9-mer库。通过跳过AbinitioRelax的'Relax'阶段以减少计算时间,在CASP11、CASP12和CASP13测试集中的每个自由建模蛋白质靶标生成了20,000个蛋白质模型。通过TM-score48评估这些采样结构模型的质量。
3. 结果讨论
现有片段文库从蛋白质数据集中获得候选片段。相比之下,DeepFragLib在模板片段数据库(HR956)中仅使用956个高质量链进行库构建,并且在获得的片段和构建的模型中实现了最佳性能。仅使用高质量结构可避免与低质量结构相关的噪声和误差,有利于深度神经网络的训练,而不会损害模型的普遍性。
DeepFragLib的整体层次结构旨在降低计算复杂性并提高计算效率。粗粒度CLA模型在每个位置仅保留> 200,000个碎片中的前5,000个作为细粒度REG模型的输入,这反过来简化了超深度REG模型的训练和推断。单独的CLA模型无法提供准确的片段识别。在CASP13集中,仅由CLA模型选择的顶部片段构建的控制片段库表现出所有度量的显著性能降低。这突出了REG模型在检测近原生片段中的重要性。串联连接分类和回归模块允许有效地招募高质量片段,因为从所有片段数据(不经过CLA模型过滤)对REG模型的训练和推断在计算上是禁止的。
从LRFragLib到DeepFragLib,引入了对片段库组成的两个主要更改。首先,将最大片段长度从10个延伸到15个残基,因为较长的片段可以提高蛋白质折叠模拟中的采样效率,因为这些片段包括更多的非局部结构偏好。其次,与贪婪浓缩步骤中可以招募无限数量片段的LRFragLib不同,DeepFragLib中的片段数量在每个位置限制在50到200之间。LRFragLib倾向于在特定位置获得更多碎片,特别是在螺旋区域,因此,螺旋,支架和线圈碎片的部分是不平衡的。将片段编号限制为[50,200]后,DeepFragLib中的不平衡会得到缓解。
Rosetta结构预测中,NNMake意外地在DeepFragLib之后产生了第二好的性能,而Flib-Coevo产生的结果与其片段库的质量评估中的更高精度不一致。因此,除精确度和覆盖范围之外的其他因素也可能有助于结构预测。一个可能的因素是“深度”,即预测片段的平均数量。
参考资料
Wang T, Qiao Y, Ding W, et al. Improved fragment sampling for ab initio protein structure prediction using deep neural networks[J]. Nature Machine Intelligence, 2019, 1(8): 347-355.
Data availability
https://doi.org/10.24433/CO.3579011.v1
Code availability
https://github.com/ElwynWang/DeepFragLib
https://doi.org/10.24433/CO.3579011.v1
http://structpred.life.tsinghua.edu.cn/DeepFragLib.html