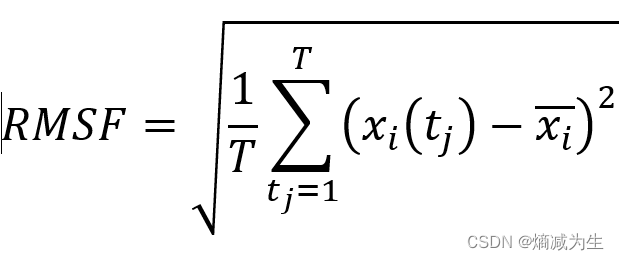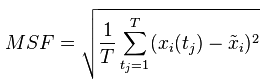欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://blog.csdn.net/caroline_wendy/article/details/129125467
参考文档:Nature Methods | 蛋白、RNA、DNA及其复合物结构的比对算法US-align
官网地址:https://zhanggroup.org/US-align/
TMScore
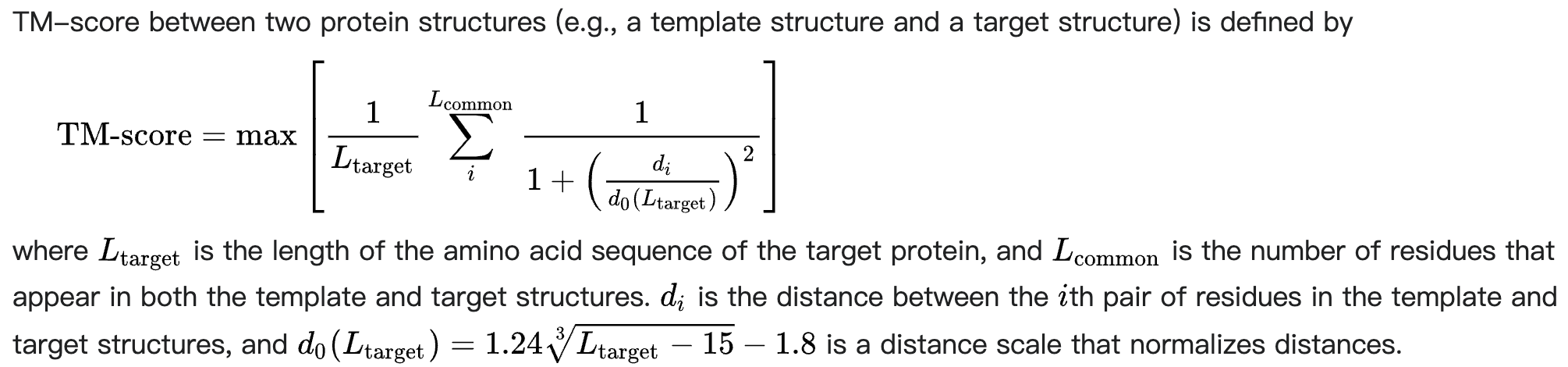
TMScore,即 Template Modeling Score,参考Wikipedia、ZhangGroup:
US-align (Universal Structural alignment) is a unified protocol to compare 3D structures of different macromolecules (proteins, RNAs and DNAs) in different forms (monomers, oligomers and heterocomplexes) for both pairwise and multiple structure alignments.
- US-align(通用结构比对)是一种统一协议,用于比较不同形式(单体、低聚物和异源复合物)的不同大分子(蛋白质、RNA 和 DNA)的 3D 结构,用于成对和多结构比对。
The core alogrithm of US-align is extended from TM-align and generates optimal structural alignments by maximizing TM-score of compared strucures through heuristic dynamic programming iterations.
- US-align 的核心算法是从TM-align扩展而来的,通过最大化TM-score来生成最优结构比对通过启发式动态编程迭代比较结构。
Large-scale benchmark tests showed that US-align can generate more accurate structural alignments with significantly reduced CPU time, compared to the state-of-the-art methods developed for specific structural alignment tasks.
- 大规模基准测试表明,与为特定结构对齐任务开发的最先进方法相比,US-align 可以生成更准确的结构对齐,同时显着减少 CPU 时间。
TM-score has values in (0,1] with 1 indicating an identical structure match, where a TM-score ≥0.5 (or 0.45) means the structures share the same global topology for proteins (or RNAs).
- TM-score 的值在 (0,1] 范围内,其中 1 表示相同的结构匹配,其中 TM-score ≥0.5(或 0.45)表示结构共享相同的蛋白质(或 RNA)全局拓扑结构。
TMScore的计算,一般使用US-align,来源于
- US-align: Universal Structure Alignment of Proteins, Nucleic Acids and Macromolecular Complexes
- 蛋白质、核酸和大分子复合物的通用结构比对
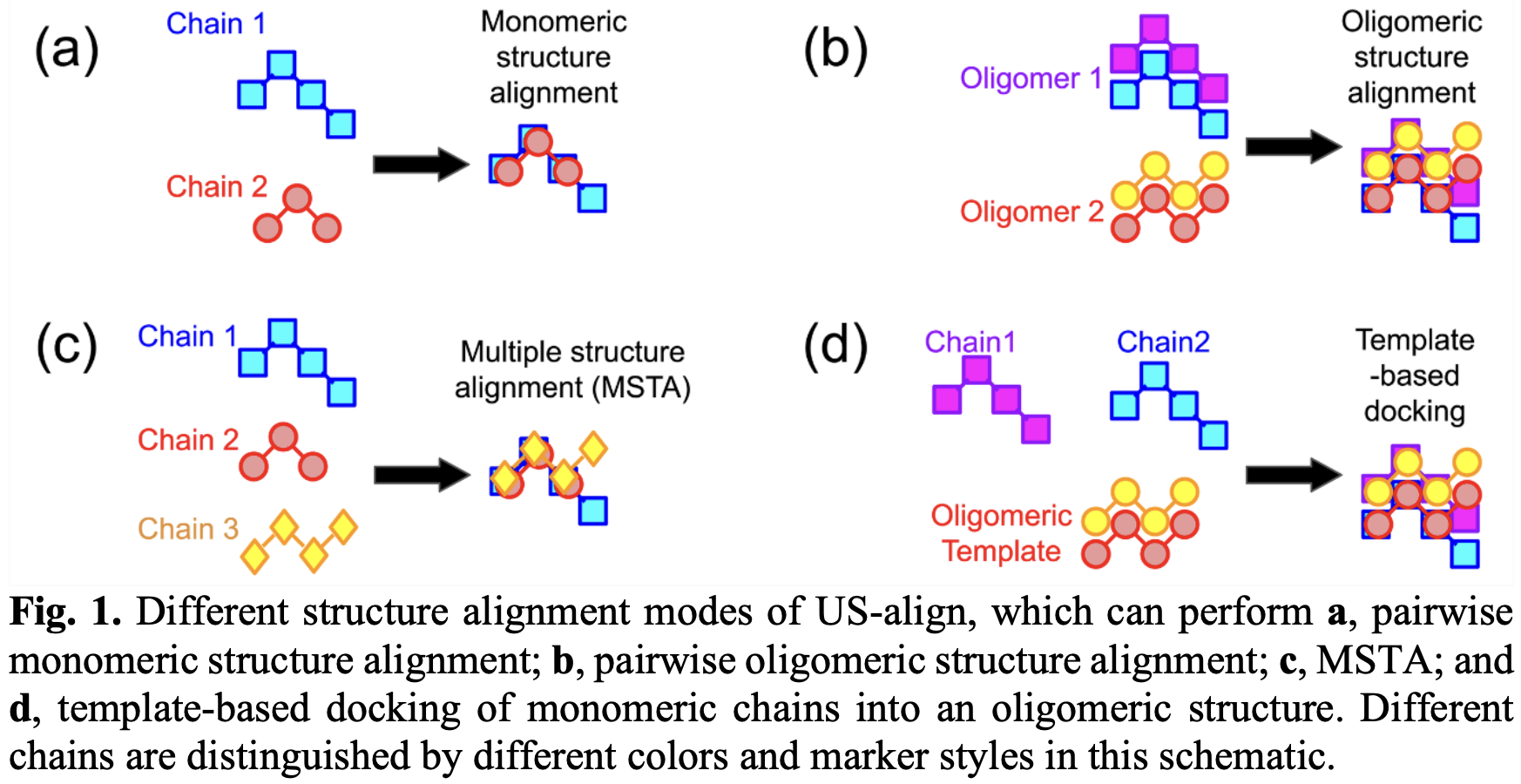
全部比对功能都统一使用的TM-score作为打分函数。支持:
- 单链结构的两两比对,Monomeric Structure Alignment
- 复合物(低聚物)结构的两两比对,Oligomeric Structure Alignment
- 多结构比对,Multiple Structure Alignment,MSTA
- 基于模板的分子对接,Template-based Docking
RMSD
Root Mean Squared Deviation,均方根偏移,是重叠后的两个蛋白质的原子(一般是主链原子)之间的平均距离的度量 。完全重合为0A,一般认为小于3A时,两个结构相似。
计算公式:
R M S D ( X , X r e f ) = min R , t 1 N ∑ i = 1 N [ ( R ⋅ X i ( t ) + t ) − X i r e f ] 2 RMSD(X,X^{ref})=\min_{R,t}\sqrt{\frac{1}{N}\sum_{i=1}^{N}[(R \cdot X_{i}(t)+t)-X^{ref}_{i}]^2} RMSD(X,Xref)=R,tminN1i=1∑N[(R⋅Xi(t)+t)−Xiref]2
- X X X和 X r e f X^{ref} Xref为两个要对比的蛋白, X i X_i Xi和 X i r e f X_{i}^{ref} Xiref为用于比对的两个原子, N N N为比对的序列长度
- 蛋白重叠对齐的过程就是,找到一组旋转矩阵R和向量t使得两个用于比较的蛋白 X X X和 X r e f X^{ref} Xref有最小的RMSD
具体步骤:
- 下载USalign.cpp:https://zhanggroup.org/US-align/bin/module/USalign.cpp
- 编译CPP脚本:
g++ -static -O3 -ffast-math -lm -o USalign USalign.cpp- macOS,不使用’-static’,即
g++ -O3 -ffast-math -lm -o USalign USalign.cpp
注意:如果编译TMalign.cpp,无法编译,将#include <malloc.h>替换为#include <stdlib.h>
源码:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2022. All rights reserved.
Created by C. L. Wang on 2023/2/16
"""import os
import subprocessfrom root_dir import DATA_DIR, ROOT_DIRclass TMScoreAndRMSD(object):"""TMScore,来源于USalign"""def __init__(self):self.bin_dir = os.path.join(ROOT_DIR, "bin")self.usalign = os.path.join(self.bin_dir, "USalign")def get_tms_and_rmsd_with_usalign(self, pdb1_path="", pdb2_path=""):"""计算 TM Score参考: https://zhanggroup.org/US-align/"""if not pdb1_path or not pdb2_path:return -1.0cmd = " ".join([f"{self.usalign}", "-TMscore 1", "-outfmt 2", pdb1_path, pdb2_path])res = subprocess.Popen(cmd, shell=True, stdout=subprocess.PIPE).stdout.read()res = res.decode("utf-8")# [PDBchain1 PDBchain2 TM1 TM2 RMSD ID1 ID2 IDali L1 L2 Lali]# IDali 一致ID/对齐ID# Lali 对齐长度items = res.split("\n")[1].split("\t")tm_score = (float(items[2]) + float(items[3])) * 0.5 # TM1和TM2的均值rmsd = float(items[4])return tm_score, rmsddef main():pdb1_path = os.path.join(DATA_DIR, "test_7roa.pdb")pdb2_path = os.path.join(DATA_DIR, "7roa.pdb")tsu = TMScoreAndRMSD()tm_score, rmsd = tsu.get_tms_and_rmsd_with_usalign(pdb1_path, pdb2_path)print(f"[Info] tm_score: {tm_score}, rmsd: {rmsd}")if __name__ == '__main__':main()
Sequence
标准FASTA序列,例如7ROA:
- https://www.rcsb.org/structure/7ROA
>7ROA_1|Chain A|EntV|Enterococcus faecalis OG1RF (474186)
SDQLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDASTVVQARFNWNALGSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCGLS源码:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2022. All rights reserved.
Created by C. L. Wang on 2023/2/17
"""
import os.path
import warningsfrom Bio.Data.PDBData import protein_letters_3to1_extended as d3to1
from Bio.PDB import PDBParserfrom root_dir import DATA_DIRdef get_seq_from_pdb(pdb_path):"""从PDB中获取序列:param pdb_path: PDB路径:return: PDB结构"""# 常见的20种结构# d3to1 = {'CYS': 'C', 'ASP': 'D', 'SER': 'S', 'GLN': 'Q', 'LYS': 'K',# 'ILE': 'I', 'PRO': 'P', 'THR': 'T', 'PHE': 'F', 'ASN': 'N',# 'GLY': 'G', 'HIS': 'H', 'LEU': 'L', 'ARG': 'R', 'TRP': 'W',# 'ALA': 'A', 'VAL': 'V', 'GLU': 'E', 'TYR': 'Y', 'MET': 'M',}record = pdb_path # PDB路径# 解析器parser = PDBParser(QUIET=True)structure = parser.get_structure('struct', record)res_str = ""for model in structure:for chain in model:header = chain.idseq = []for residue in chain:if residue.resname in d3to1.keys():seq.append(d3to1[residue.resname])elif residue.resname == "HOH":continueelse:warnings.warn(f"{residue.resname} don't support!")seq_str = "".join(seq)res_str += f">{header}\n{seq_str}\n"return res_strdef main():pdb_path = os.path.join(DATA_DIR, "7roa.pdb")res_str = get_seq_from_pdb(pdb_path)print(f"[Info] res: \n{res_str}")if __name__ == '__main__':main()