转载于 https://mp.weixin.qq.com/s/VDN1qAZGIMol6prwQW4umw
Docking非原生配体
在前面的例子中,AutoDock Vina能把配体构象调整到几乎原生的构象,验证了这一预测方法的准确度。下面,我们尝试docking另外一个配体药物nelfinavir奈非那韦,来展示如何寻找小分子在蛋白内的结合位点。这个过程可以进一步地凝练和扩展作为“虚拟筛选(virtual screening)”的步骤。
重复上述步骤执行docking
-
获取nelfinavir.pdb:为教程提供的pdb文件(可从
1OHR.pdb获得) -
按照上述步骤对配体文件进行预处理获得
pdbqt格式文件。 -
修改配置文件,执行Docking,输出日志如下,并用
PyMOL可视化结果。Detected 4 CPUs Reading input ... done. Setting up the scoring function ... done. Analyzing the binding site ... done. Using random seed: 2009 Performing search ... done. Refining results ... done.mode | affinity | dist from best mode| (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+----------1 -11.2 0.000 0.0002 -11.0 1.878 9.6183 -9.8 1.354 4.2544 -9.6 1.732 8.6795 -9.5 1.192 1.8146 -9.2 1.669 2.2697 -9.0 2.003 8.0758 -8.7 1.850 3.8039 -8.4 1.856 9.549 Writing output ... done.
评估docking结果
-
对这个例子来讲,PDB中存在nelfinavir与HIV-1蛋白酶的晶体结构(1OHR),可以作为金标准来检测docking的准确性。
-
PyMOL中导入
1OHR.pdb文件,在对象面板中依次点选1OHR行-H-Hide everything-S-Show cartoons-C-By chain。从图中可以看到这两个蛋白酶体在空间的方向不同,因此我们需要重新比对这两个结构,运行PyMOL> align 1OHR, 1hsg_prot,可以看到两个结构完全重合了。You may have observed that moving the structure around the window is a bit difficult since the origin of the view has been altered when you loaded 1OHR.pdb. To reset it, try:
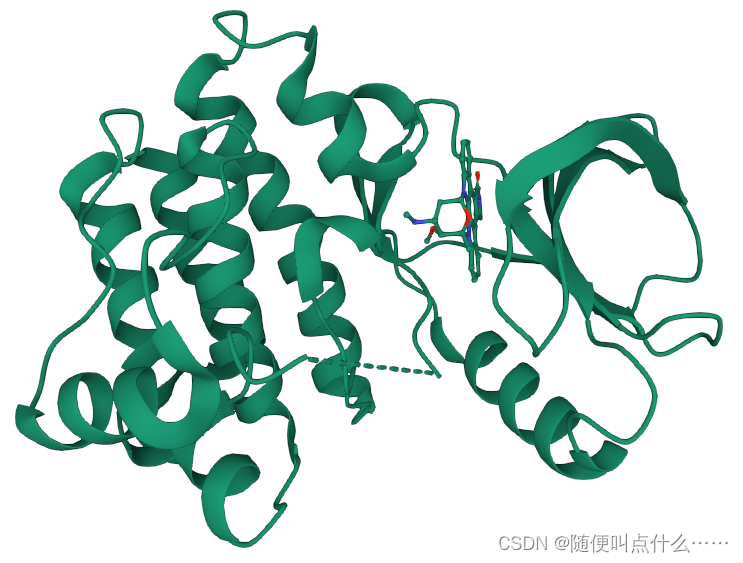
PyMOL> reset,运行之后没有看到变化。Docking结果展示。第一张图表示2个晶体结构align前的展示;第二张图表示2个晶体align后重合在了一起。白色化合物为1OHR PDB晶体结构中配体nelfinavir的构象,视为金标准。红色为本教程的结果(只加极性氢)。
-
展示PDB文件中的蛋白结合的化合物提取
1OHR中的nelfinavir (残基为1UN),运行PyMOL> select nelfinavir, 1OHR and resn 1UN;在对象面板更改其展示方式,依次点选S-Show sticks-C-white。通过与金标准比对,判断哪个构象是预测的最佳模式。Docking第一张图表示AutoDock Vina输出结果的Best Mode与金标准的比对情况;第二张图表示AutoDock Vina输出结果的Second Best Mode与金标准的比对情况;白色化合物为1OHR PDB晶体结构中配体nelfinavir的构象,视为金标准。红色为本教程的结果(只加极性氢)。
结果看到
second best mode看上去吻合的更好,为什么呢?从日志的结合能量来看,best mode和second best mode只差了0.2。那么还有一个问题,1SHG的chainA与1OHR的chainA是不是一个呢?我们比对1OHR的chainA与1HSG的chainB,
PyMOL> align 1OHR and chain A, 1hsg_prot and chain B。Docking结果展示。第一张图表示2个晶体结构align后重合在了一起。第二张图表示1OHR的chainA与1HSG的chainB比对的结果。白色化合物为1OHR PDB晶体结构中配体nelfinavir的构象,视为金标准。红色为本教程的预测的second best mode结果(只加极性氢)。
用PyMOL在蛋白表面搭建静电层 (electrostatic surface)
静电作用在分子docking过程中发挥着重要的作用,接下来将观察静电力是如何与配体作用的。前面提到,PDB结构中不包含原子的局部电荷信息,而这对静电力场的计算是很重要的。因此我们需要给PDB文件中增加这一数据。
为了完成这一任务,我们需要在http://www.poissonboltzmann.org/注册,然 后下载安装软件APBS和pdb2pqr。
-
在Windows下,
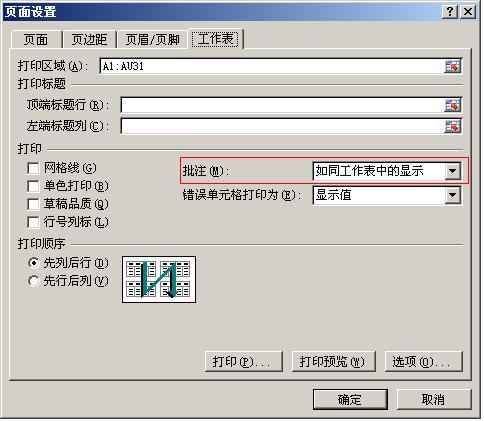
APBS直接下载使用默认的安装目录安装即可;pdb2pqr解压缩到C:\pdb2pqr; 路径中不能有空格。设置环境变量:
我的电脑-属性-高级系统设置-高级-环境变量-系统变量中选中PATH-编辑-新建-加入安装路径(如下图所示)安装完成之后,启动PyMOL,会在
Plugin下看到APBS Tools。 -
在Lunux下,尚未试验。
打开PyMOL并读入1hsg_prot.pdb,然后通过下述步骤启动并配置APBS, 依次点击菜单或按钮:
-
Plugin-APBS Tools-Main-Select Use PyMOL generated PQR and PyMOL generated Hydrogens and termini(这步操作是给PDB文件中的每个原子 加氢、局部电荷和计算原子半径;This adds hydrogens and assigns partial charges and atomic radii to each atom in the PDB file.) -
Configuration-Set grid(点击后定义了一个保护蛋白的框,但并未显示 ,因此点击后看不到框,但可以看到一系列的计算过程体现在展示界面。This defines a grid that encloses the protein, but Grid is not displayed on the screen) -System Temperature = 300-on concentration (+1) and (-1) to 0.15(相当于 0.15摩尔的阳离子和阴离子, which is equivalent of 0.15M cations and anions) -
按图设置APBS和pdb2pqr的路径 -
Run APBS-Visualization-Update(如果出现Unable to open file error,运行命令PyMOL > load C:\Users\ct\AppData\Local\Temp\pymol-generated.dx) -Molecular Surface - Show
左图为配置加氢的参数;中图是设置GRID;右图为设置可执行文件的路径
左图是展示APBS计算结果;中图为计算结果路径;右图为结果展示
静电等高线图(Electrostatic isocontours)
PyMOL makes this step very easy: adjust the positive and negative “Contour” fields to the desired values (usually ±1, ±5, or ±10 kT/e) and hit the Positive Isosurface and Negative Isosurface and Show buttons.
±1 kT/e electrostatic potential isocontours of FAS2 in PyMOL
If the colors are not as you expect, you can change the colors of the objects iso_neg and iso_pos in the main menu. By convention (for electrostatics in chemistry), red is negative (think oxygen atoms in carboxyl groups) and blue positive (think nitrogen atoms in amines).
得到这个图之后,我们首先需要看配体是否落在受体的”口袋”里;然后检查配体与受体之间原子的化学匹配,如配体中的碳原子应该与受体的疏水原子结合, 氮原子和氧原子与其受体中相近原子结合;然后看有没有电荷互补;最后根据已有知识查看结合q区域有没有包括蛋白的活性位点, 以及活性位点怎么与受体相互作用的。
用ADT可视化结果
-
导入Vina输出结果:打开ADT依次点选
Analyze-Dockings-Open AutoDock Vina result-选择`结果PDBQT文件dockingResult.pdbqt-Single molecular with multiple conformations`。 -
导入蛋白结构:
Analzye-Macromolecule-Open-1hsg_prot.pdbqt左图是Vina结果展示;右图为蛋白结果展示
-
展示相互作用:
Analyze-Dockings-Show interactionsThis display is radically different: the viewer background color is white, the ligand is displayed with a solvent-excluded molecular surface, atoms in the receptor which are hydrogen-bonded or in close-contact to atoms in the ligand are shown as spheres AND pieces of secondary structure are shown for sequences of 3 or more residues in the receptor which are interacting with the ligand. The GUI for this command lets you turn on and off different parts of this specialized display as well as list interactions in the python shell.
配体与受体作用展示, 使用方向键切换不同的配体构象
虚拟筛选
-
准备受体文件
prepare_receptor4.py -r 1hsg_prot.pdb -o 1hsg_prot.auto.pdbqt -A hydrogens。【注:脚本在目录mgltools_x86_64Linux2_1.5.6/MGLToolsPckgs/AutoDockTools/Utilities24下, 自行添加到环境变量或参照软件安装部分】 -
准备配体文件
prepare_ligand4.py -l indinavir.pdb -o indinavir.auto.pdbqt -A bonds_hydrogens。 -
还有关键一步是确定搜索空间,书写conf.txt文件。可以简单的以蛋白的中心为搜索空间的中心,蛋白各个维度坐标值的标准差、极差及其组合分别作为搜索空间的大小;在大范围搜索结束后,根据docking结果再重新选取Docking小分子的中心为搜索空间的中心,其各个维度坐标值的标准差、极差及其组合分别作为搜索空间的大小,再进行精细搜索。
-
执行Docking
vina --config conf.txt -
prepare_receptor4.py和prepare_ligand4.py支持pdb\mol2格式文件。
FAQ
-
怎么判断哪个是想要的结果?
When the results are sorted by lowest-energy, the compounds which bind as well as your positive control or better can be considered potential hits. (Remember to allow for the ~2.1 kcal/mol standard error of AutoDock). If you have no positive control, consider the compounds with the lowest energies as potential hits.)
-
怎么分析结果?
Sort them by lowest energy first, then use ADT to inspect the quality of the binding. Generally it is wise to inspect the top 30 to 50 results.
-
可视化结果时关注哪些方面?
A: The first thing to check is that the ligand is docking into some kind of pocket on the receptor. The second is that there is achemical matchbetween the atoms in the ligand and those in the receptor. For example, check that carbon atoms in the ligand are near hydrophobic atoms in the receptor while nitrogens and oxygens in the ligand are near similar atoms in the binding pocket. Check for charge complementarity. Check whatever else you may know about your particular system: for instance, if you know that the enzymatic action of your protein involves a particular residue, examine how the ligand binds to that residue. In the case of HIV protease, good inhibitors bind in a mode which mimics the transition state.
-
配体小分子获取
-
NCI Diversity Set
To expedite drug discovery, the National Cancer Institute maintains a resource of more than 140, 000 synthetic chemicals and 80, 000 natural products for which it can provide samples for high-through-put screening (HTS). The NCI Diversity Set is a collection of 1990 compounds selected to represent the structural diversity in the whole resource.
-
ZINC
ZINC Is Not Commerical is a free database of over 4.6 million commercially-available compounds for virtual screening (blaster.docking.org/zinc).
如何绘制小分子?
-
使用Gaussview从头画出配体的空间结构模型保存为mol2文件,稍微复杂的分子在画完后需要做一下量子化学水平的结构优化
-
如果配体十分复杂,可以先使用ChemDraw或ChemBioDraw画出配体结构的平面图,保存成cdx后缀的文件,然后使用OpenBabel转换成mol2文件
babel -icdx Ligand_1.cdx -omol2 ligand.mol2 --gen3D[参数--gen3D输出立体结构]。 -
SDF文件转mol2,
babel -i sdf Ligand.sdf -o mol2 ligand.mol2 --gen3D。
始终需要加氢吗?
Yes, for both the macromolecule and the ligand, you should always add hydrogens, compute Gasteiger charges and then you must merge the non-polar hydrogens. Polar hydrogens are hydrogens that are bonded to electronegative atoms like oxygen and nitrogen. Non-polar hydrogens are hydrogens bonded to carbon atoms.
可以使用AutoDock确认潜在的结合位点吗?
如果不知道配体在受体上的结合位点,就设置一个大到足够覆盖整个受体蛋白表面的长方体(在每个维度设置更多的grid points,加大grid spacing)。然后执行Docking。利用这次分子对接的结果再针对性的设定Grid的大小和位置,再执行Docking。如果蛋白特别大,那么可以分多次设置Grid,如第一次覆盖蛋白上面2/3, 第二次覆盖中间2/3,第三次覆盖下面2/3等。
确定大分子活性位点方法总结
-
在PyMOL中, 载入两个蛋白
-
用align 将未知活性位点的蛋白与配体-受体蛋白进行比对
-
标记未知活性位点的蛋白残基
-
保存比对并标记过的未知活性位点蛋白
-
查阅文献,根据文献报道找到活性位点。
-
如果有受体-配体的三维结构,则可以运用配体扩张法,确定活性位点,就是以配体的位置为中心,再向外扩增一定范围,一般为6.5到9埃,这个范围的受体残基就构成了相关的活性位点。
-
利用分子空洞技术列如MOE中的site Finder模块,然后根据经验规律,(疏水残基最多的空洞为活性位点)判断活性位点。
-
Discovery Studio Visualizer (free)观察配体结合位点,也可试试from PDB Site records或from receptor cavities确定活性位点。
-
有一个活性位点预测网上服务器 Q-Site Finder 地址http://www.modelling.leeds.ac.uk/qsitefinder/
-
找一个序列结构类似的有配体-受体复合物的3D结构,与未知活性位点的蛋白进行对比:
如果某一蛋白受体有多个晶体。我们要从中选择那一个比较好呢?:
-
采用解析晶体分辨率较高的
-
观察晶体图的B-因子
-
蛋白和配体的平均B-因子之间的不同
-
配体、受体的电子密度。
-
选择的蛋白受体的来源与研究的生物体一致
-
选择残基(特别是活性位点)完整,分辨率高的蛋白受体。
-
选择结合位点,温度系数较低的蛋白受体。
-
选择配体物与蛋白形成复合物的蛋白,最好配体的构想、结构构像与研究的小分子类似。