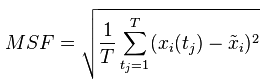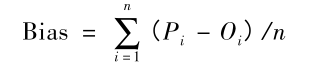参考:https://zhuanlan.zhihu.com/p/347743101
https://www.codenong.com/cs106148400/
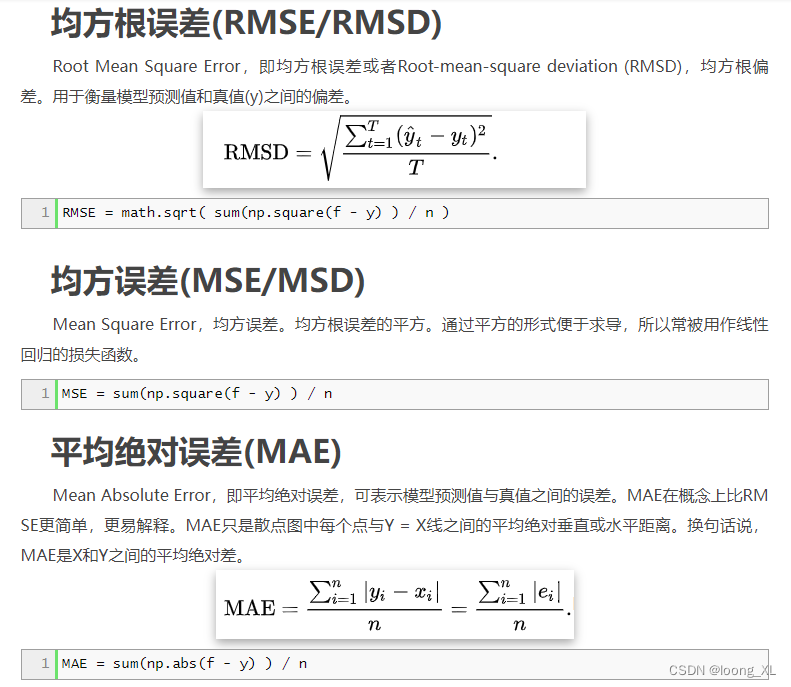
RMSD
单位是埃 RMSD,root-mean-square deviation,也就是均方根偏差。 原子位置的均方根偏差是叠加蛋白质的原子(通常是骨架原子)之间的平均距离的量度。注意,RMSD计算可以应用于其他非蛋白质分子,如小的有机分子。在球状蛋白质构象的研究中,通常在刚体进行完叠加后通过计算Cα原子坐标之间的RMSD来表征三维结构的相似性。 等式:
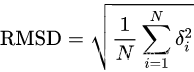
通常,RMSD用作两种或更多种蛋白质结构之间相似性的定量测量,通常越低越好。
命令行
#rank1, 4o75_ligand是两个分子
align rank1, 4o75_ligand

另外方法:
pymol 加载python计算代码:
参考:https://zhuanlan.zhihu.com/p/430944478
python脚本:rmsd_3d_structure.py
from pymol import cmddef shortest_permol(mol_1, mol_2):'''ref: mol_1compute rmsd of mol_2'''mol1_model = cmd.get_model(mol_1)mol2_model = cmd.get_model(mol_2)num = 0lst_atom_dis = []dis_shorest = 1000for a in mol2_model.atom:# num += 1for b in mol1_model.atom:# print("model %s and index %d"%(mol_2, a.index),"model %s and index %d"%(mol_1, b.index))dis = cmd.get_distance("model %s and index %d"%(mol_2, a.index), "model %s and index %d"%(mol_1, b.index))if dis_shorest > dis:dis_shorest = dislst_atom_dis.append(dis_shorest)num += dis_shorestreturn num/len(lst_atom_dis)def rmsd_three(mol_1, mol_2):refmol1_commol2 = shortest_permol(mol_1, mol_2)refmol2_commol1 = shortest_permol(mol_2, mol_1)rmsd = round((refmol1_commol2 + refmol2_commol1)/2,2)# print(refmol1_commol2, refmol2_commol1)print("The rmsd of " + mol_1 + " between " + mol_2 + " is " + str(rmsd))return rmsdcmd.extend("rmsd_three", rmsd_three)
pymol加载使用:
1、cd 到脚本目录,比如我这放在桌面上的
注意脚本目录地址不要有中文
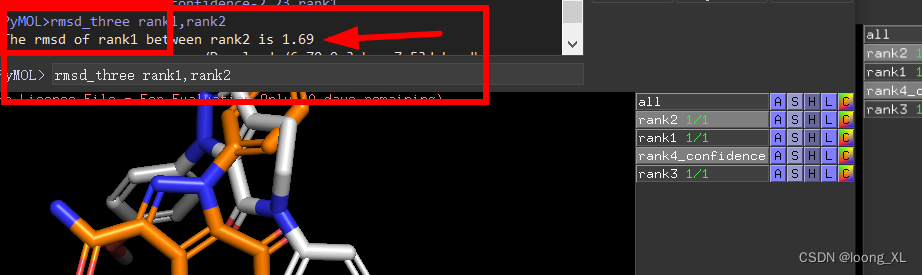
2、run脚本:run rmsd_3d_structure.py

3、使用rmsd命令:rmsd_three rank1,rank3
2、spyrmsd库计算RMSD
参考:https://spyrmsd.readthedocs.io/en/develop/tutorials/tutorial.html#loading-molecules
安装:pip install spyrmsd
##单独两个分子
from spyrmsd import io, rmsdref = io.loadmol(r"C:\***\1au3_B_PCM.sdf")
dock_ = io.loadmol(r"***\rank4_confidence-0.78.sdf")coords_ref = ref.coordinates
anum_ref = ref.atomicnums
adj_ref = ref.adjacency_matrixcoords_dock_ = dock_.coordinates
anum_dock_ = dock_.atomicnums
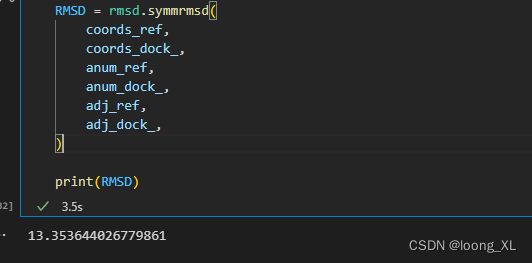
adj_dock_ = dock_.adjacency_matrixRMSD = rmsd.symmrmsd(coords_ref,coords_dock_,anum_ref,anum_dock_,adj_ref,adj_dock_,
)print(RMSD)
##多个分子计算
ref = io.loadmol("molecules/1a4k_ligand.sdf")
mols = io.loadallmols("molecules/1a4k_dock.sdf")coords_ref = ref.coordinates
anum_ref = ref.atomicnums
adj_ref = ref.adjacency_matrixcoords = [mol.coordinates for mol in mols]
anum = mols[0].atomicnums
adj = mols[0].adjacency_matrixRMSD = rmsd.symmrmsd(coords_ref,coords,anum_ref,anum,adj_ref,adj,
)print(RMSD)
RMSE\MSE\MAE区别