最近在讲课演示的时候,偶尔会发生如下的异常。“配置系统未能初始化”,当时也没有时间深究到底什么原因造成的。


今天再一琢磨,以上的InnerException提示说:无法识别的配置节userSettings。

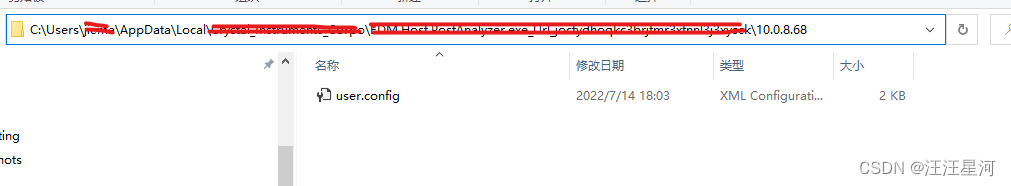
再一深入找下去,我们找到下面这个目录

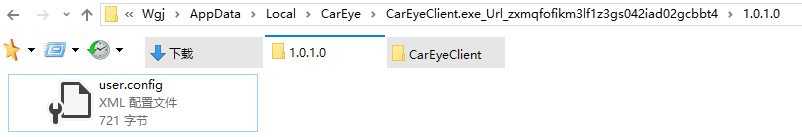
这个目录下面确实有一个user.config文件

这个文件内容是

这是之前我做另外一个程序时保存的用户配置文件。
所以,问题找到了。因为我前后两个应用程序的名称是一样的。虽然现在这个程序没有需要userSettings,但它仍然试图去读取上述提到的user.config文件。当然就出错了
所以,解决方案是:将C:\Documents and Settings\ChenXizhang\Local Settings\Application Data目录下面的WindowsFormsApplication1目录删除即可