点击蓝字 关注我们
短读长扩增子测序是否适用于微生物组功能的预测?

https://doi.org/10.1002/imt2.38
PERSPECTIVE
●2022年7月4日,巴西圣保罗医院肿瘤分子诊断中心Vitor Heidrich教授和德国朱利叶斯库恩研究所Lukas Beule教授在iMeta在线发表了题为“Are short-read amplicons suitable for the prediction of microbiome functional potential? A critical perspective”的文章。
● 该研究阐述了利用分类组成(特别是来源于短读长测序时)预测微生物组功能之生物学可信度方面的一些担忧,讨论了标记基因的分类学分辨率、标记基因的基因组内变异以及微生物组数据的组成性质。
● 共同第一/通讯作者:
Vitor Heidrich (vheidrich@mochsl.org.br);
Lukas Beule (lukas.beule@julius-kuehn.de)
摘 要
标记基因的分类学分析可以较低成本揭示微生物群落的分类概况,所以在微生物组研究中使用频率较高。现在有越来越多的工具可以从这类数据中提取更多的生物信息。在这篇观点中,我们阐述了利用分类组成(特别是来源于短读长测序时)预测微生物组功能之生物学可信度方面的一些担忧,讨论了标记基因的分类学分辨率、标记基因的基因组内变异以及微生物组数据的组成性质。将微生物组功能的实际测定与预测相结合可以更好地理解微生物组功能。文中我们强调了功能预测对产生和验证假设的重要性。我们认为,在提取微生物组DNA的基础上进行短读长扩增子测序预测微生物组功能不应作为生物学推断的唯一基准。
关键词:组成数据,功能潜力组成,标记基因的基因组内变异性,微生物功能,微生物组数据,短读长扩增子测序,分类学分辨率
亮 点
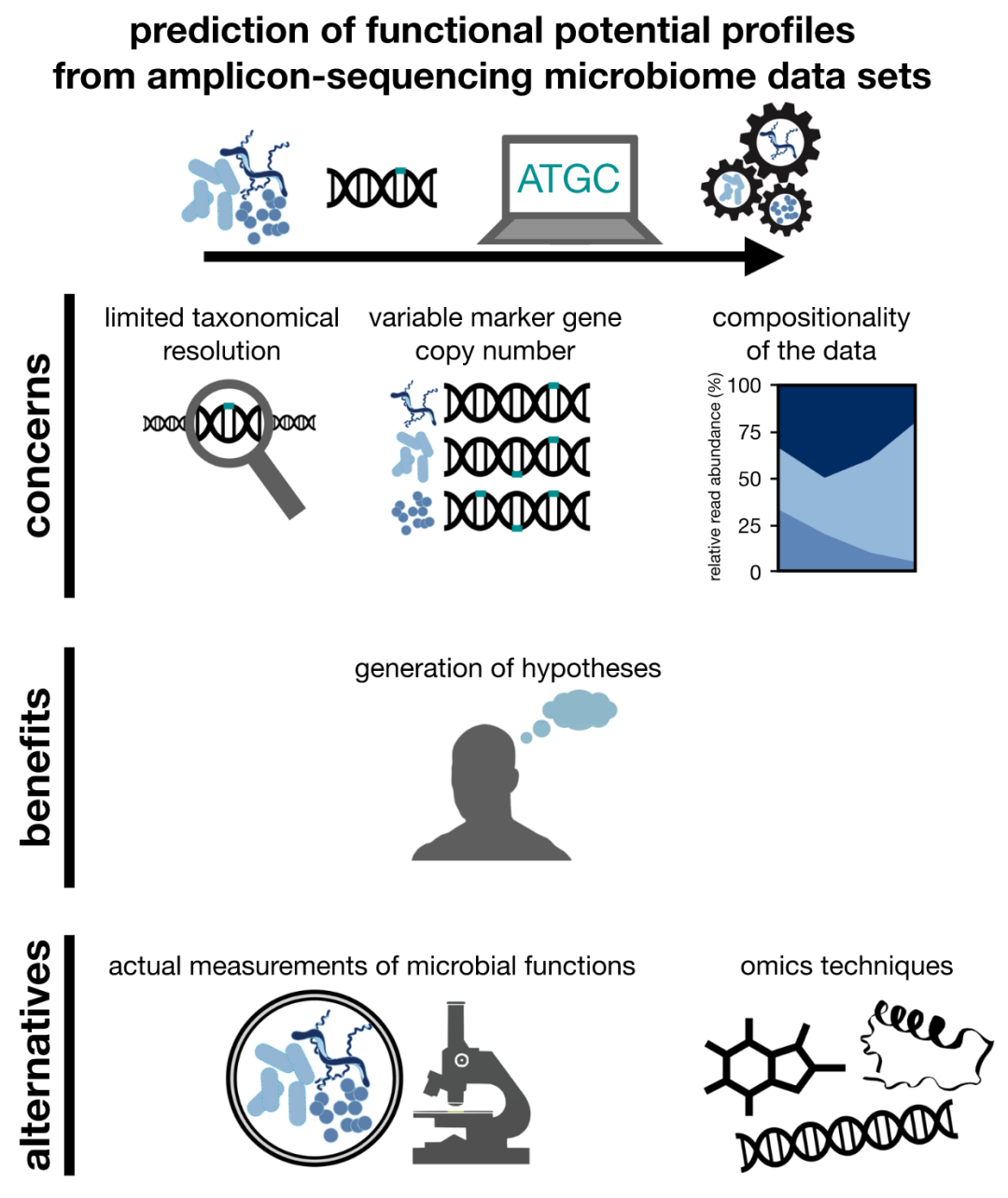
● 阐述了利用微生物组数据集分类谱预测微生物潜在功能之生物学可信度方面的一些问题
● 讨论了标记基因的分类学分辨率、标记基因的基因组内变异以及微生物组数据的组成性质
● 将微生物组功能的实际测定与预测相结合是理解微生物组功能的一个好办法
全文解读
引 言
在过去的20年里,二代测序(NGS)技术广泛用于微生物群落分析。现在的NGS平台价格实惠,允许用户同时测试大批量样本,并具有较高的碱基读取精度。这些技术使研究人员能够破译不同生境微生物群落的组成(e.g., [1–3])。随着微生物组研究的迅速发展,出现了一些可以从微生物组数据集的分类情况预测功能潜力概况的工具[4]。微生物潜在功能谱通常列出微生物组中可能会存在的微生物功能,并根据预测的微生物丰度揭示相应的微生物功能的相对重要性。微生物功能对理解微生物群落很重要,而这些工具可以补充微生物组分类学数据集。尽管这些工具提供了许多额外的信息,在这篇文章中我们就使用短读长扩增子测序产生的微生物组数据集的分类注释来推导微生物功能表达了一些担忧。虽然预测算法本身已经讨论得很深入(e.g., [4]),我们关注的是短读长扩增子测序数据预测微生物功能的适用性,而不是预测算法的效率。
扩增子测序的分类学分辨率
在下笔时,使用Illumina平台的短读长扩增子测序是最常见的测序技术。在Illumina广泛使用的MiSeq系统上,短序列的双端测序可以产生多达2×300个碱基对。在微生物分析中,短读长扩增子测序最常用的对象是具有分类学信息的细菌16S核糖体RNA (rRNA)和真菌内转录间隔子(ITS)基因位点,但这些基因的部分测序往往在种水平的区分度不高。例如,大约20年前,Blackwood et al.[5]对芽孢杆菌(Bacillus spp.)16S rRNA基因的高可变V1-V3区域进行了测序,发现无法区分两种临床相关的蜡样芽孢杆菌(Bacillus cereus)类群——炭疽芽孢杆菌(B. anthracis)和蜡样芽孢杆菌(B. cereus)。事实上,对蜡样芽孢杆菌组16S rRNA全基因的测序显示,只有部分炭疽芽孢杆菌和蜡样芽孢杆菌具有可分辨的16S rRNA序列[6, 7],指出该标记基因对某些类群的分类学分辨率有限。最近,人们指出了菌株水平鉴定人类微生物组组成的重要性[8, 9],然而短读长扩增子测序大多无法区分相关度高的菌株[9]。
已有研究证明了一个不言自明的事实,采用PacBio等长读长测序技术可以实现16S全长测序,提高了分类学分辨率(e.g., [10])。作为长读长测序的替代方案,Loop Genomics提出通过采用Illumina短读长测序技术,结合在每个16S扩增子独特的标签序列来合成长读长序列,也可以在种水平上提高分辨率[11]。此外,通过使用诸如SMURF[12]这样的计算工具整合针对不同16S高变区域的扩增子测序结果,可以推断出具有更高分辨率的分类学图谱。这种替代方法目前更容易使用,因为不需要替换测序平台或调整建库准备工作流程。然而,这不是最优的选择,因为与Loop Genomics技术相反,全长16S序列无法用这种策略重建。
尽管16S全长测序广泛应用的成本仍然高昂,但我们预测,从长远来看,通过长读长扩增子测序或基于唯一识别的短读长扩增子测序的生物信息学重建实现的16S全长测序将逐渐取代短读长测序,提高了分类学分辨率,进而更准确地预测微生物的潜在功能。
除了分类学上的区别之外,同一种水平的菌株之间的基因变异进一步阻碍了功能的准确预测。2004年,Jaspers和Overmann发现11株Brevundimonas alba具有完全相同的16S rRNA基因序列,但具有较高的基因组多样性和生理学特征差异性[13],表明需要分离微生物才能精确评估它们的功能。虽然公共数据库中缺乏参考基因组无疑限制了功能预测工具的作用发挥,但这个例子进一步说明,参考基因组可用并不代表功能推断可靠。
标记基因的基因组内变异性
利用扩增子测序数据分析微生物组结构的另一个难点是微生物基因组中标记基因的拷贝数有差异。真菌可以包含多达几百个被ITS分隔开的rRNA基因拷贝,但又在不同的真菌中呈现数量级的差异[14]。即使在同一种水平的真菌菌株中,各基因组的18S和28S rRNA基因的拷贝数也可以有很大的差异[14–17]。Lavrinienko等人[18]推测,ITS等非转录区域可能具有更大的拷贝数变异性。同样,如Rainey 等人[19]研究所示,细菌可能含有多拷贝的16S rRNA基因。最近的估计表明,每个细菌物种16S rRNA基因拷贝数的中位数可以在1到19之间变化[20]。虽然现在已经开发出了修正古细菌和细菌基因组中16S rRNA基因拷贝数的工具[21,22],但基因拷贝数修正的可靠性仍存在不确定性和争议[23,24]。此外,从本世纪之初人们已经知道,单个细菌基因组中的16S rRNA基因并不总是相同的(例如[25])。这可能会导致扩增子测序把本属于同一细菌细胞中的16S rRNA等位基因认定为不同的细菌种类[26],从而会高估多样性[27]。因此,除了可变标记基因拷贝数在试图评价所研究环境中每个种类的相对贡献时的明显含义(即相对丰度失真)外,这种变异性有时会认为是等位基因多样性,甚至会混淆微生物组的组成。由于潜在功能谱反映了分类谱中的所有偏差,标记基因的基因组内变异性会干扰潜在功能的相对重要性估计,进而可能会高估潜在功能谱的多样性。
微生物组数据的组成性质
由于NGS从样本中产生的序列读数与样本中的细菌细胞数量之间没有关系[28],因此这个读数并不转化为细菌丰度。NGS中每个分类单元产生的读数仅反映了群落一部分的相对大小,使NGS微生物组数据集是可组装的[29]。换句话说,这意味着它解锁了一个微生物群落中类群的相对测序读取丰度(即比例或频率),但由于整个群落的大小(微生物生物量)仍然未知,它没能揭示类群的绝对丰度[28–31]。因此,即使一个群落的预测功能谱与其实际功能相吻合,但由于未考虑该群落的总种群规模,而无法对功能潜力的大小进行估计。例如,假定有定两个组成相同的微生物组(微生物组A和微生物组B),且微生物组A的种群大小是B的两倍,我们希望比较它们的预测功能潜力和实际功能潜力。虽然它们的预测功能潜力是相同的,但实际上微生物组A的功能潜力是B的两倍。目前也有很多其他的微生物定量方法可以克服微生物组分析的这一局限性(如[28,30,32–34]),但是不同的量化方法可能会引入额外的数据异质性。
另一种规避微生物组成数据分析问题的策略是使用比率[36],因为使用比率可以消除微生物自身负荷造成的偏差[37]。所以如果微生物群的功能可以用分子/功能/过程比率来表示,正如生物系统中的碳氮比、白蛋白/球蛋白比、中性粒细胞/淋巴细胞比一样,潜在功能的相对丰度变成了有价值的信息,微生物的生物量变得不那么关键。虽然不总是实用,且我们对数据组成特性本身的担忧依然存在,但我们建议在分析功能谱时尽可能使用比率。无论如何,使用比率的成分感知统计方法使数据不太依赖于微生物生物量,因此这类方法也是很有用的,应该得到青睐[29]。
最后,尽管这里没有讨论,但值得一提的是,PCR偏差是一个众所周知的误差来源,它可以曲解群落组成(例如[38])和功能预测.
测定微生物群的实际功能
另一种要求更高的方法是在尽量测定微生物群的实际功能。比如,受环境微生物调控的过程(如酶活性[39]、温室气体排放通量[40]和固氮[41])通常可以原位测定。同样地,通过正交测定粪便代谢物,可以评估人体/动物肠道微生物的功能,这已经被成功地用于研究肠道微生物和生物转化的关系,这些关系一定程度上可解释一些包括免疫抑制剂在内的药物[42-43]在病人体内的差异性。只有当微生物群落可获取,在采样时相应的功能是活跃的,并且有足够的种群大小和采样量用于检测,测量微生物的实际功能才是可行的。对于不满足这些标准的微生物群,使用实时荧光定量PCR等额外技术是对某类功能基因遗传潜能定量表征的一种有意义的补充。但必须指出的是,这些遗传潜能并不一定转化为微生物活动和过程[44–46]。从这个角度来说,转录组学、蛋白质组学和代谢组学等组学技术有助于探索已经表达的遗传潜能。我们认为,结合功能预测和特定功能的实际测定是了解微生物功能的一个强有力的方法。
产生和验证假设
尽管有局限性,但我们承认功能预测有产生新假设的特殊潜力。然而,我们尤其不能忽视微生物群落是复杂的。因此,功能组成是高维的,很难分析。这意味着通常潜在功能预测工具会引发太多的研究方向,而产生直接的假设变得困难。我们强烈鼓励研究人员选择有意义的假设,并尽可能独立地检验这些假设。例如Zhang 等人[47]预测了小鼠肠道菌群的代谢功能,并通过核磁共振代谢组学成功验证了他们的预测。同样,Wu等人[48]预测了结直肠腺瘤和结直肠癌患者肠道微生物组相比健康个体发生了生物合成途径的改变,且通过实时荧光定量PCR定量测定这些途径中的基因验证了他们的预测。尽管有关验证预测功能的研究是个例而不是规律,但这些例子说明了潜在功能预测在发现和探索新的研究方向方面的优势。
总 结
本文中所讨论的微生物组功能潜能预测的问题、益处和替代方案见图1。总的来说,我们肯定那些为实现微生物组分类学数据集功能预测所做的努力,也坚信功能预测有助于产生新的思路和潜在研究方向。但我们认为,在提取微生物组DNA的基础上进行短读长扩增子测序预测微生物组功能不应作为生物学推断的唯一基准,而从短读到长读测序技术的转变将有助于克服这些问题。尽管如此,高分辨率的分类分析并不能解决我们提出的关键问题(例如,微生物组数据的组成性质)。因此,在功能预测的同时,结合使用组学(如代谢组学)和非组学方法(如qPCR)表征微生物活性对阐明微生物组功能至关重要。
引文格式:
Vitor Heidrich, Lukas Beule. 2022. Are short-read amplicons suitable for the prediction of microbiome functional potential? A critical perspective. iMeta 1: e38. https://doi.org/10.1002/imt2.38
更多推荐
(▼ 点击跳转)
iMeta文章中文翻译+视频解读
iMeta封面 | 宏蛋白质组学分析一站式工具集iMetaLab Suite(加拿大渥太华大学Figeys组)
▸▸▸▸
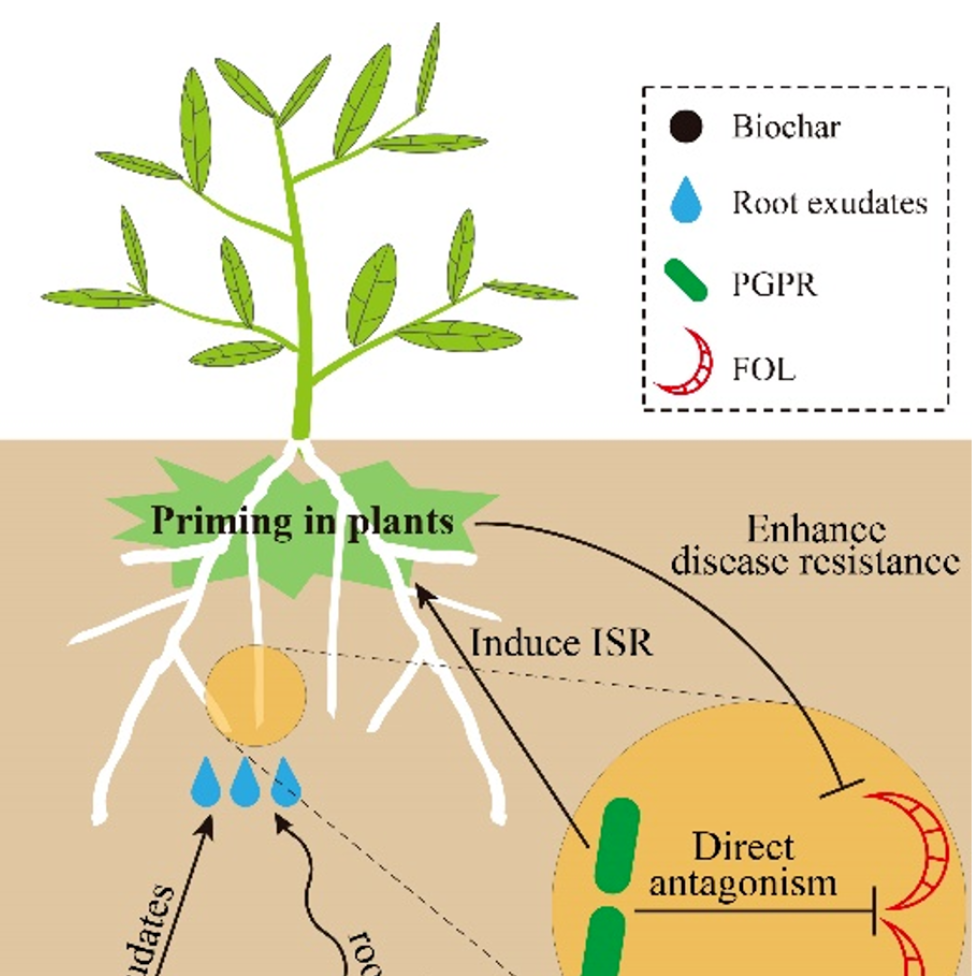
iMeta | 东农吴凤芝/南农韦中等揭示生物炭抑制作物土传病害机理
▸▸▸▸
iMeta | 华南农大陈程杰/夏瑞等发布TBtools构造Circos图的简单方法
▸▸▸▸
iMeta | 叶茂/时玉等综述环境微生物组中胞内与胞外基因的动态穿梭与生态功能
▸▸▸▸
iMeta | 南农沈其荣团队发布微生物网络分析和可视化R包ggClusterNet

▸▸▸▸
iMeta | 华南师大王璋组综述人体肺部微生物组与人类健康和疾病之间的隐秘关联
▸▸▸▸
iMeta | 南科大夏雨组纳米孔测序揭示微生物可减轻高海拔冻土温室气体排放
▸▸▸▸
iMeta | 北大陈峰/陈智滨等发表口腔微生物组研究中各部位取样的实验方法(Protocol)
▸▸▸▸
iMeta | 华南农大曾振灵/熊文广等-家庭中宠物犬与主人耐药基因的共存研究
▸▸▸▸
iMeta | 深圳先进院马迎飞组开发基于神经网络分析肠道菌群的方法
▸▸▸▸
iMeta | 南医大陈连民等综述从基因组功能角度揭示肠菌对复杂疾病的潜在影响
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
微信公众号
iMeta
责任编辑
微微