点击蓝字 关注我们
易扩增子(EasyAmplicon):微生物组研究中易用的扩增子分析流程

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.83
● 2023年1月27日,中国农科院基因组所刘永鑫团队、中国中医医科学院陈同团队和南京农业大学文涛团队等在 iMeta 在线发表了题为 “EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research ” 的文章。
● 本研究提供了一个跨平台、开源和社区支持的分析流程——易扩增子(EasyAmplicon)。易扩增子包括30多个跨平台模块和该领域常用的R包。流程由文章作者和“宏基因组”公众号编辑团队维护和更新,定期发布最新的中英文教程,阅读用户的反馈,并在微信公众号和GitHub中为用户提供帮助。该流程可在 GitHub (https://github.com/YongxinLiu/EasyAmplicon) 和 Gitee (https://gitee.com/YongxinLiu/EasyAmplicon)上获得。
● 第一作者:刘永鑫、陈雷、马腾飞
● 通讯作者:陈同 (chent@nrc.ac.cn)、文涛(taowen@njau.edu.cn)、刘永鑫(liuyongxin@caas.cn)
● 合作作者:李小方、郑茂盛、周欣、陈亮、钱旭波、席娇、卢洪叶、曹慧荦、马晓亚、边遍、张鹏帆、吴季秋、甘人友、贾保磊、孙林阳、鞠志成、高云云
● 主要单位:中国农业科学院深圳农业基因组研究所、首都医科大学附属复兴医院、兰州大学、中国科学院遗传与发育生物学研究所农业资源中心、华北电力大学、中国科学院微生物研究所、浙江大学附属金华医院、西北农业科技大学资源与环境学院、浙江大学口腔医院、香港大学、泰国皇太后大学、日本东京大学、德国马普植物育种研究所、爱尔兰科克大学、荷兰格罗宁根大学、新加坡食品与生物技术创新研究所、韩国中央大学、南京农业大学、中国中医医科学院中药资源中心
亮 点
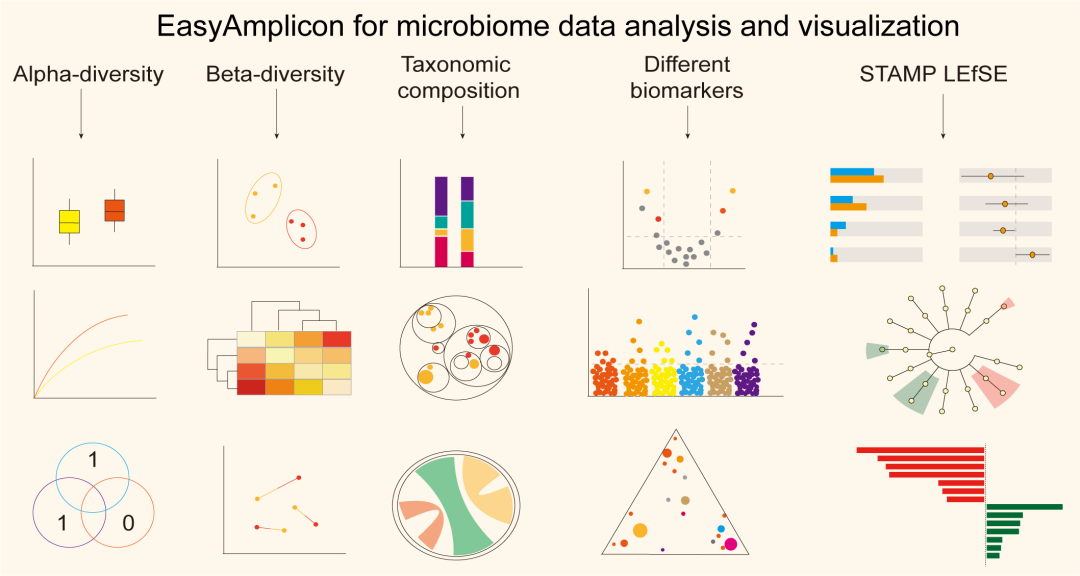
● 易扩增子 (EasyAmplicon) 是一个用户友好的、跨平台的、社区支持的扩增子数据分析流程
● 拥有微生物组研究中数据处理和可视化的大部分模块
● 流程定期维护和更新,并且鼓励用户贡献自己的分析代码
摘 要
初学者学习和使用扩增子分析软件比较困难,因为可供选择的软件工具太多,而且都需要很多步骤。我们提供了一个跨平台、开源和社区支持的分析流程——易扩增子(EasyAmplicon)。易扩增子拥有扩增子分析所需的大部分模块,包括数据质量控制、双端读取合并、去重复、聚类或去噪、嵌合体检测、特征表生成、物种多样性分析、分类组成、生物标志物发现和出版级图表。易扩增子包括30多个跨平台模块和该领域常用的R包。流程所有步骤的运行都可在RStudio中使用,降低学习成本,保持分析过程的灵活性,便于个性化分析。流程由文章作者和“宏基因组”公众号编辑团队维护和更新,定期发布最新的中英文教程,阅读用户的反馈,并在微信公众号和GitHub中为用户提供帮助。流程可以部署在各种平台上,安装时间不到半小时。在普通笔记本电脑上,3小时内即可完成几十个样品的全部分析过程。该流程可在GitHub(https://github.com/YongxinLiu/EasyAmplicon)和Gitee (https://gitee.com/YongxinLiu/EasyAmplicon)上获得。
视频解读
Bilibili:https://www.bilibili.com/video/BV1T8411c7oa/
Youtube:https://youtu.be/M1AdE5B9GJQ
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
近二十年来高通量测序技术的快速发展,促进了微生物组在人类、动物、植物和环境中核心功能的探索越来越深入。他们中的大多数发现是由扩增子测序驱动的(例如细菌或古细菌的16S rDNA 测序、真核生物的18S rDNA或内部转录间隔区,以及固氮原核生物的nifH 基因),并分析了各种环境中微生物组的物种组成。
扩增子测序的湿实验室操作现已标准化,大部分操作由专业的生物技术公司或测序中心执行。然而,扩增子数据的生物信息学分析仍然具有挑战性,大量的软件、方法和算法的存在给初学者的使用选择带来了很大困难。主流的扩增子分析流程包括 mothur、USEARCH和QIIME ,它们都被引用了10,000 多次。然而,它们仍然存在明显的缺点,如缺乏下游统计分析和可视化解决方案、学习时间成本较高、受限于特定操作系统等。一些在线分析网络服务器易于使用,例如 Qiita 、MGnify 和 gcMeta ,但它们也有一些问题,例如上传速度慢、等待/运行时间长、可调参数少,这使得无法进行定制分析 。
缺乏易用和灵活的扩增子分析流程严重限制了研究人员对数据分析过程的理解,阻碍了该领域的发展。因此,我们开发了一个易用、开源和跨平台的扩增子分析流程——易扩增子(EasyAmplicon)。它可以在 RStudio 中以命令行模式和交互模式使用。目前,它提供了20多种可视化样式,可以轻松生成出版级质量的图表。开源代码可以促进可重现的分析并允许个性化修改。此外,它还为最流行的软件生成标准输入,例如 STAMP 、LEfSe 、PICRUSt 1 & 2 、BugBase 、FAPROTAX 、ImageGP 和iTOL 。易扩增子为扩增子分析提供了一个免费、可重复和个性化的解决方案,这可能是微生物组研究的一个很好的软件工具。
结 果
易扩增子 (EasyAmplicon) 流程概述
易扩增子是适合于在笔记本电脑或服务器上进行扩增子数据分析和可视化的整合分析流程,可提供各种表格和可视化结果来探索潜在的生物学解释。此流程易于安装在Windows、MacOS和Linux 系统上。安装方法在“Methods”部分有详细介绍或可在https://github.com/YongxinLiu/EasyAmplicon 上获取。测试数据包含 18 个样本且每个样本 包含50,000 对 PE250 读长的短测序序列,完整的分析可以在大约3小时内完成,峰值内存占用量小于4 GB(CPU:2 核,2.1 GHz)。
易扩增子是一个端到端的分析流程。它从原始读长开始、输出数据表和出版质量图片(图 1)。主要包括降维、分析、统计可视化三个步骤(图1)。所有相关软件都易于安装(表 1),我们提供批量下载包来加速流程部署。
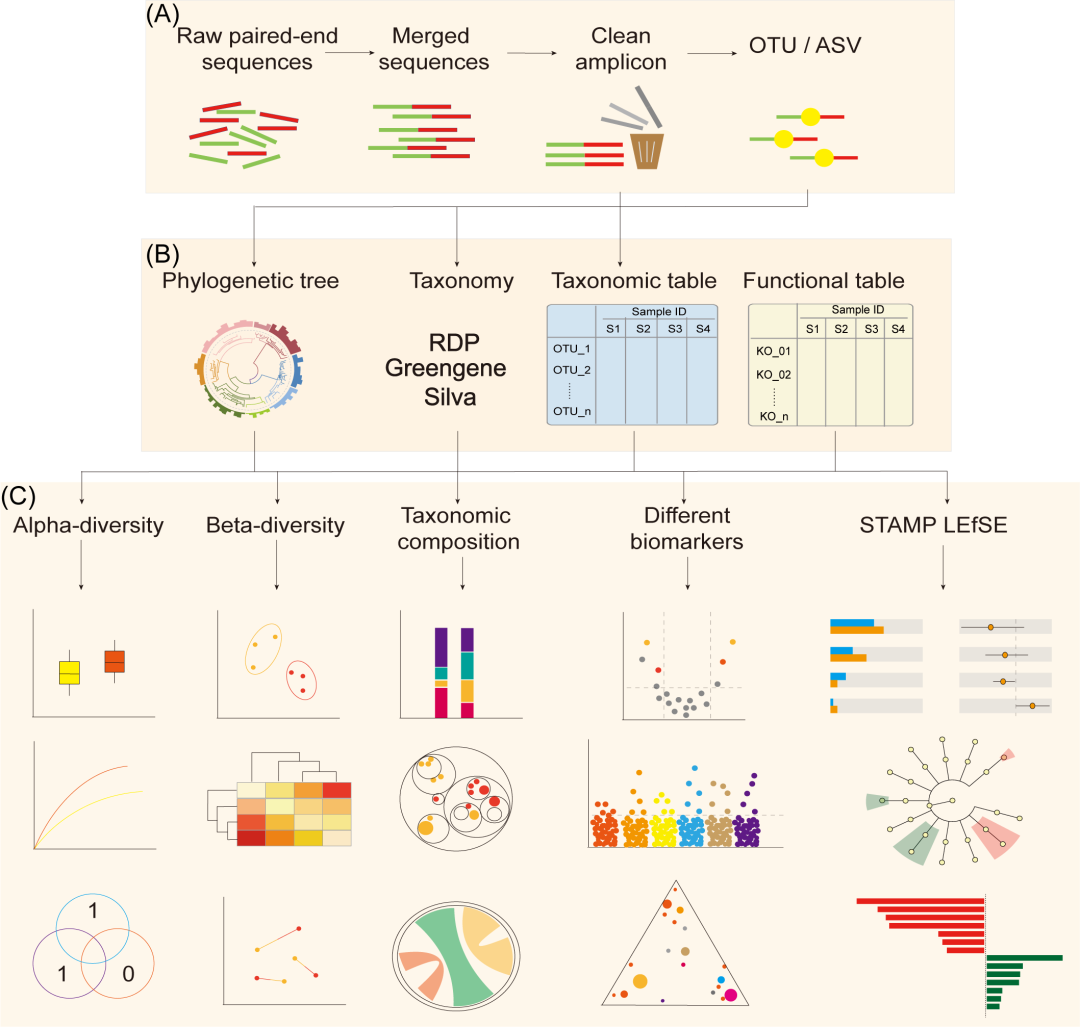
图1. 用于分析双端扩增子序列的易扩增子流程
(A) 降维:将原始测序读长处理成特征表。(B) 分析:提供系统发育分析、物种分类、功能预测以及 alpha 和 beta 多样性计算。(C) 统计可视化:生成出版质量的图表并为生物学解释进行统计测试。
表 1. 易扩增子中包括的软件和包

在命令行或 R markdown 模式下运行流程
首先,我们使用 RStudio 打开流程文件“pipeline.sh”。设置好工作目录后,只需用鼠标点击“运行”按钮,即可一步步运行分析过程。用户进行自己的分析,只需要原始测序数据和样本元数据,易扩增子会处理后续分析。如果 RStudio不可用,我们可以将脚本复制并粘贴到 pipeline.sh 中,然后在任何 Shell 环境(例如本地或远程 Linux/Mac 中的终端,或 Windows 的 Git bash)中运行它们。所有相关软件和包详见表1所示。所有图形默认保存为PDF格式,部分示例见图2和图3所示。
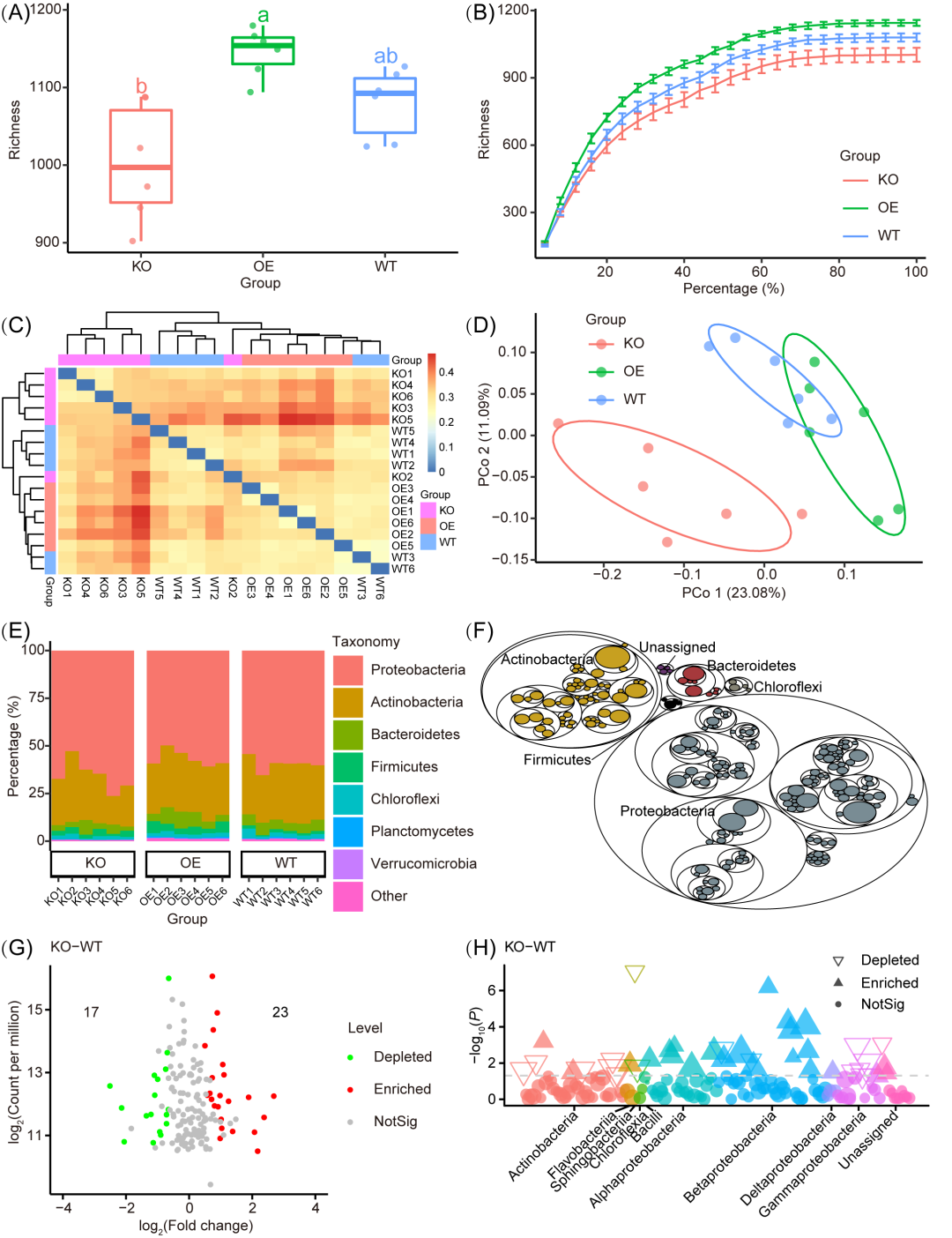
图2. 出版级可视化示例
(A) 箱线图显示组间丰富度指标的 alpha 多样性。不同的字母表示显著不同的组(P < 0.05,ANOVA,Tukey HSD)。方框内的水平条代表中位数。方框的顶部和底部分别代表第75个和第25个百分位数。上部和下部延长线分别从框的上边缘和下边缘延伸到不超过四分位距 1.5 倍的数据。(B) 丰富度的稀疏曲线表明,随着测序深度的增加,特征达到饱和阶段。每个垂直条代表标准误差。(C) 基于Bray-Curtis距离的样品聚类热图。(D) 基于Bray-Curtis 距离的主坐标分析 (PCoA)。(E) 门级分组样本中物种组成的堆积条形图。(F) 分类组成的树图。(G) 火山图显示 KO 和 WT 组之间存在显著差异的丰度分类群。(H) 曼哈顿图显示了 KO 和 WT 组之间的不同特征和相关分类群。此图中样本重复的数量如下:敲除(KO,n = 6),过表达(OE,n = 6)和野生型(WT,n = 6)。
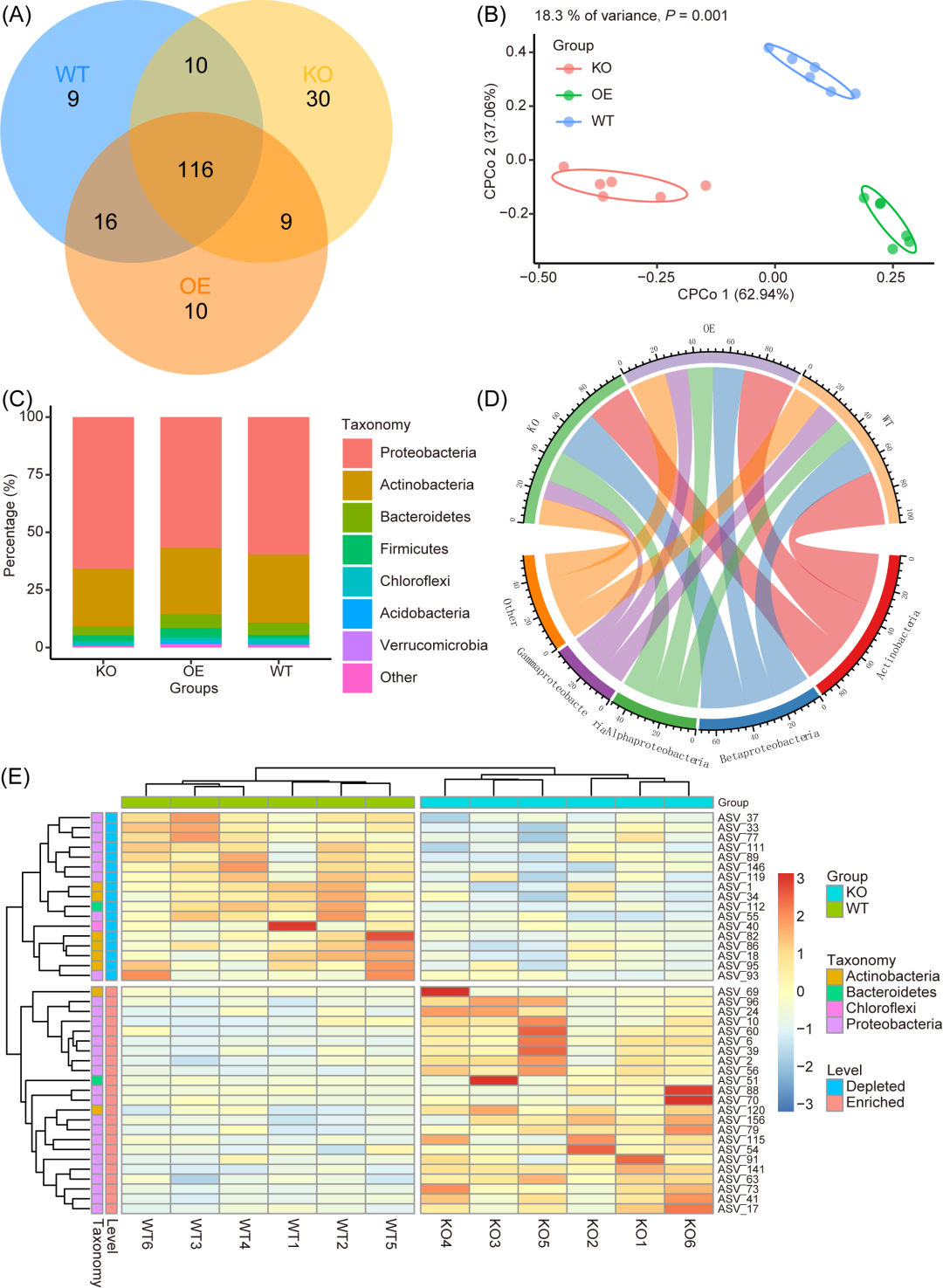
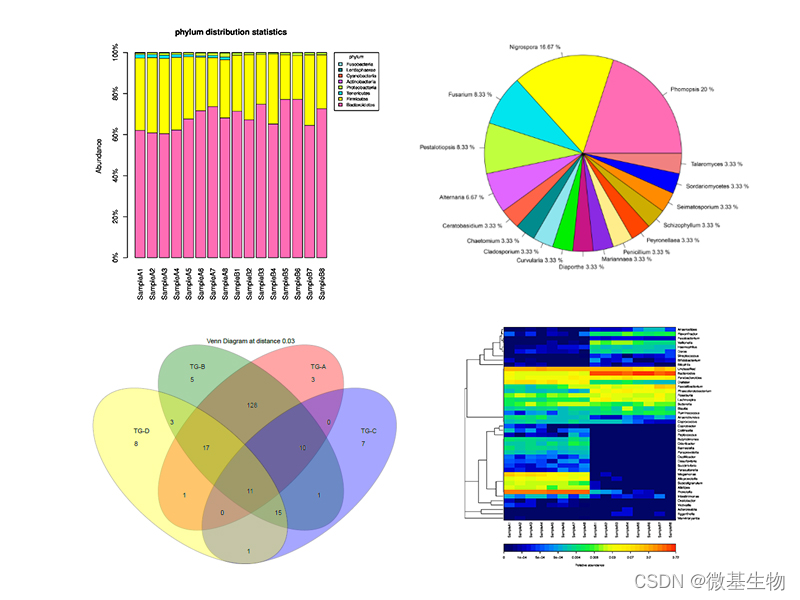
图3. 对图 2 中的出版物质量可视化的补充示例
(A) 维恩图显示三组中共有和独特的 ASV(相对丰度 > 0.1%)。(B) 三组的约束性主坐标分析 (CPCoA)。(C) 三组门级别平均相对丰度的堆叠图。(D) 三组门级平均相对丰度的圈图。(E) 热图显示 KO 组和 WT 组之间显著差异的 ASV(Wilcoxon 检验,P < 0.05)。
为了使微生物组数据的统计和可视化更加个性化,用户可以在RStudio中打开“Tutorial.Rmd”文档,然后修改图形的细节,如分组顺序、配色方案、图例布局等。它甚至可以生成可发表的组合图(图2和3)和可重现的 HTML 格式报告(Tutorial.html)。
第三方软件支持
易扩增子并未涵盖微生物组分析所需的所有功能。目前有一些主流且非常有特色的微生物组分析工具,如STAMP 、LEfSe 、PICRUSt 1 & 2 、BugBase 、FAPROTAX 和iTOL 。然而,一些输入文件对于没有生物信息学背景的用户来说很难准备。在 易扩增子中,大量脚本用于为上述软件准备输入,并轻松生成示例可视化,如STAMP(图 4A)、LEfSe(图 4B)、BugBase(图 4C)和 iTOL(图 4D)。至于最流行的 QIIME 2 流水线,易扩增子生成的中间文件可以导入 QIIME 2,QIIME 2 的输出文件也可以导入易扩增子进行下游分析。
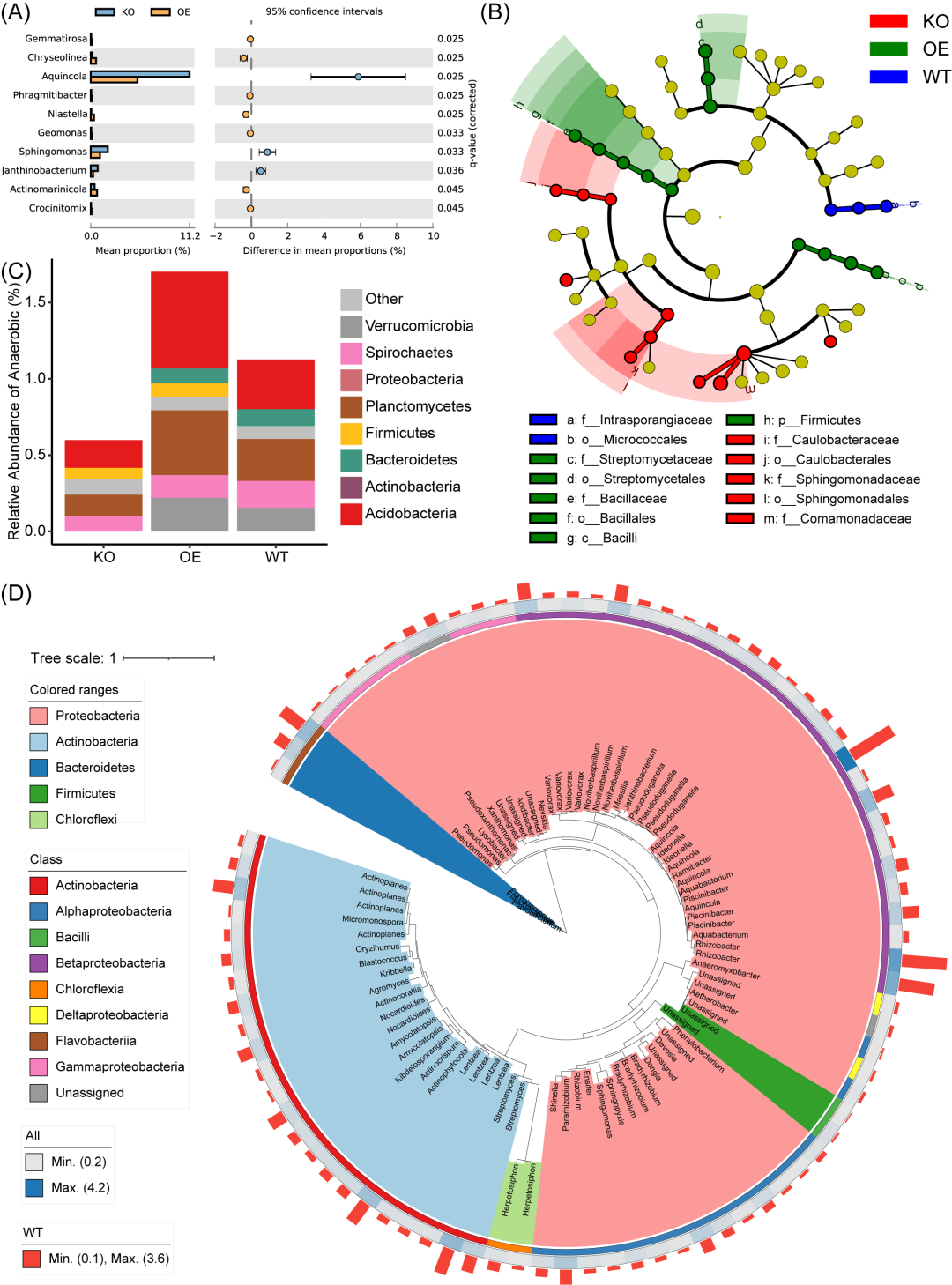
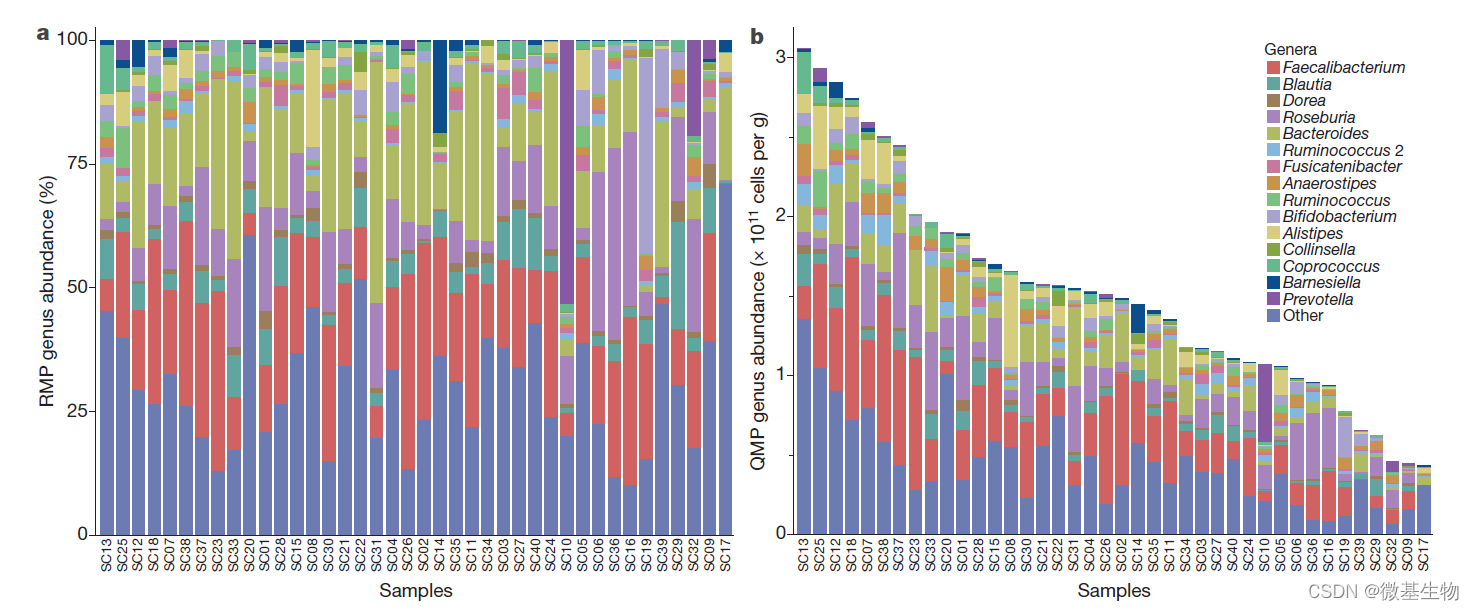
图4. 第三方软件使用易扩增子中间文件生成的可视化结果
(A) STAMP在WT和KO组中属级的扩展柱状图。(B) 通过LEfSe显示每组生物标志物的进化分支图。(C) BugBase注释厌氧菌在门级的百分比。(D) 86个ASV的系统发育树(相对丰度>0.2%)。树的背景按Phylum着色。外条代表不同的纲。热图表示所有样本的平均相对丰度。条形图表示ASV 在WT组的相对丰度。
预期结果
易扩增子为扩增子数据分析提供多种可视化样式。对于 alpha 多样性(样本内多样性),箱线图是可视化数据和比较每组异同的最佳方式(图 2A),不同的字母代表显著差异(P < 0.05,ANOVA,Tukey HSD 检验)。稀释曲线分析表明,随着测序深度的增加,特征达到饱和阶段,线条和误差条分别代表均值和标准误(图 2B)。如果您想检查样本或组之间的独特或共同特征,维恩图是显示此模式的好方法(图 3A)。至于 beta 多样性,基于 Bray-Curtis 距离的热图将是一种很好的可视化方法。彩色分组标签显示样本聚类与哪些属性相关(图 2C)。
讨 论
目前,对于扩增子分析,最流行的流程是QIIME 和QIIME 2 ,它们已被引用 54,900次(Google Scholar 截至 2023 年 1 月 4 日)。然而,这两种流程都存在一些缺点,限制了它们在微生物组分析中的使用,例如安装包太大、不支持 Windows 系统以及缺乏出版质量的可视化。易扩增子正试图解决上述问题。
目前,这只是易扩增子的第一个版本。运行时间和内存使用取决于数据集的大小。当前版本已被数千名用户使用,截至 2022 年底正式引用 36 次(在 Google 学术搜索“EasyAmplicon”)。本文作者和“宏基因组”微信公众号核心团队将及时更新流程。相关性、网络分析 、随机森林 、机器学习 、深度学习 、迁移学习和溯源分析的脚本正在开发中,并将很快纳入分析流程。将来会建立像 MicrobiomeAnalyst这样的网络服务器版本。更通用的命令行脚本和可视化样式仍在开发中,它们将在新版本的流程中可用。欢迎任何对此项目感兴趣的人贡献有关分析方法、可视化样式或 GitHub 存储库中提到的其他问题的脚本。
结 论
总之,易扩增子流程为扩增子分析提供了一个高效的跨平台框架。此外,还提供了20多个预定义的分析和可视化解决方案,用于对数据进行多维探索并生成出版质量的图表。此外,易扩增子还提供了一些实用程序,可与其他广泛使用的软件整合使用以满足各种需求。总之,对于有或没有编程背景的研究人员来说,这套流程将极大地促进扩增子研究。易扩增子预期将每季度更新一次,以满足微生物组领域快速发展的需求。
方 法
快速使用易扩增子
易扩增子主要采用 Shell bash 和 R 语言编写,可以在命令行(终端)模式或 RStudio交互模式下运行。特别是对于没有编程知识和技能的研究人员,建议部署在 Windows 系统上(安装 Git for Windows)并在 RStudio 中运行。此外,它还支持 MacOS 和 Linux。要安装它,请按照 https://github.com/YongxinLiu/EasyAmplicon 上的说明进行操作。一些依赖的软件和包如表1所示,均己经整合以方便安装。分析过程主要包括三个步骤,如图1所示。为了证明其实用性,我们提供了一个演示数据集,其中包含属于三组的 18 个样本,每个样本稀释到 50,000 个读长。这个示例数据集是我们之前发布的数据 (CRA001464) 的一个子集(精简的示例数据存放在GSA https://ngdc.cncb.ac.cn/gsa/ 中,索引号CRA002352)。
降维 (从序列到表)
接受的输入包括双端或单端/合并序列(fastq 格式)、干净的扩增子(fasta 格式),甚至其他流程生成的中间文件,如图 1 所示。大多数扩增子在 Illumina HiSeq2500/NovaSeq6000平台上测序并采用双端 250-bp 模式。通常,流程从fastq 格式的双端读长开始,并将它们合并以获得单端序列。引物和条形码被切割,然后过滤掉低质量的读数以获得干净的扩增子。这些步骤主要使用VSEARCH或USEARCH 执行(图 1A)。16S rDNA 的干净扩增子可以直接比对到参考数据库GreenGenes 并生成特征 (OTUs) 表,它可以作为PICRUSt的输入来预测潜在的功能,并可作为 BugBase 的输入进行表型预测。另一种选择,干净的扩增子通常聚类成OTU(97% 相似性)或以从头模式去噪成扩增子序列变体 (ASV)。最后,干净的扩增子将比对到从头识别的 OTU/ASV 以生成特征表。代表性序列可用于构建系统发育树并执行物种注释(图 1A)。
分析(从大表到小表)
特征表是降维步骤的里程碑输出。我们可以使用特征表和系统发育树来计算各种alpha和 beta多样性指标。带有分类注释的特征表可用于汇总到特定的分类级别并发现所有分类级别的生物标志物(图 1B)。此外,EasyAmplicon 提供了许多“胶水脚本”来为其他广泛使用的工具生成输入文件,例如 QIIME 2 、STAMP 和 LEfSe 。
统计和可视化 (从表到图)
易扩增子可以生成 alpha 多样性、beta 多样性、物种组成和生物标志物的可视化文件以及相关统计表(图 1C,图2;表 2),这些出版质量的图表包括箱线图、散点图、堆积条形图和热图。此外,EasyAmplicon 的输出可以导入 STAMP 或 LEfSe 进行生物标志物识别,并分别可视化为扩展误差条形图或进化分枝图。
表2. 易扩增子中主要可视化功能概述

致 谢
这项工作得到了国家自然科学基金 (U21A20182) 和中国科学院青年创新促进会 (2021092) 的资助。
数据可用
原始数据存储于国家生物信息中心(GSA: CRA002352)https://ngdc.cncb.ac.cn/gsa。软件和教程详见 GitHub https://github.com/YongxinLiu/EasyAmplicon 或Gitee https://gitee.com/YongxinLiu/EasyAmplicon。
引文格式:
Yong-Xin Liu, Lei Chen, Tengfei Ma, Xiaofang Li, Maosheng Zheng, Xin Zhou, Liang Chen, Xubo Qian, Jiao Xi, Hongye Lu, Huiluo Cao, Xiaoya Ma, Bian Bian, Pengfan Zhang, Jiqiu Wu, Ren-You Gan, Baolei Jia, Linyang Sun, Zhicheng Ju, Yunyun Gao, Tao Wen, Tong Chen. 2023. EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta 2: e83. https://doi.org/10.1002/imt2.83
作者简介

刘永鑫(第一/通讯作者)
● 中国农科院基因组所研究员,iMeta期刊执行主编,宏基因组公众号创始人
● 主要研究方向为微生物组研究方法、功能研究和科学传播,在Science、iMeta、Nature Biotechnology、Nature Microbiology、Cell Host & Microbe等期刊发表论文50余篇,被引10000余次。主编《微生物组实验手册》专著,由300多位同行参与,共同打造本领域长期更新的中文百科全书。创办宏基因组公众号,14万+同行关注,累计阅读量超3千万,打造本领域最具影响的科学传播平台,免费为您团队发布成果、方法、经验、招生招聘等,欢迎投稿。2021年发起iMeta期刊,打造微生物组/生物信息领域国际顶刊,解决我本领域期刊出版卡脖子问题,建立国际学术话语权体系

陈同(通讯作者)
● 博士,中国中医科学院副研究员
● 研究方向涉及高通量数据分析、生物信息工具开发、合成生物学、表观组学等,在iMeta, Cell Stem Cell (封面文章)、Nucleic Acids Research、Nature communications、Protein & Cell等高水平杂志以第一或通讯作者发表文章十余篇;开发在线绘图平台 ImageGP,使用近 60 万人次;运营有十余万人关注的微信公众号《生信宝典》,分享有1000 多篇生物信息分析原创文章、教程和视频,阅读播放千万次。联合创办iMeta期刊,现为执行主编,致力于打造微生物和生物信息领域的国产高水平杂志

文涛 (通讯作者)
● 博士,南京农业大学钟山青年研究员, iMeta 期刊青年编委,“微生信生物”公众号创始人
● 研究方向为根际微生物生态,擅长使用多组学解析土壤微生物群落过程,开发了ggClusterNet, EasyStat等R包, Easyamplicon、Easymetabolome等组学分析流程。以第一作者在iMeta、Microbiome、ISME J、Fundamental Research、 Horticulture Research、SEL、BMC plant biology等期刊发表论文10余篇
更多推荐
(▼ 点击跳转)
高引文章 ▸▸▸▸
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
▸▸▸▸
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
▸▸▸▸
iMeta | 高颜值绘图网站imageGP+视频教程合集

第1卷第1期

第1卷第2期

第1卷第3期

第1卷第4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science