写在前面:写这篇博客只是为了总结自己对扩增子分析流程的理解,加深对扩增子的映像。
扩增子分析前的准备:
软件
qiime2(2023.5)qiime2官方下载地址
###配置文件下载:
wget https://data.qiime2.org/distro/core/qiime2-2023.5-py38-linux-conda.yml
###安装:
conda env create -n qiime2-2023.5 --file qiime2-2023.5-py38-linux-conda.yml
扩增子数据和元数据(metadata)
来自公司返回的双端测序文件(.fq.gz),metadata来自个人整理的采样信息,应包括以下内容(加粗是必须项):ID,barcode,date,site,type,group,LinkerPrimerSequence,ReversePrimer,Platform, latitude,longitude等。ps:metadata包括的信息越多越好,可以用excel编辑,然后保存为txt格式。

数据导入:
## 根据metadata生成manifest文件
awk 'NR==1{print "sample-id\tforward-absolute-filepath\treverse-absolute-filepath"} \ NR>1{print $1"\t$PWD/seq/"$1"_R1.fq.gz\t$PWD/seq/"$1"_R2.fq.gz"}' \metadata.txt > manifesthead -n3 manifest
##数据导入qiime2,格式为双端33格式:
qiime tools import \--type 'SampleData[PairedEndSequencesWithQuality]' \--input-path manifest \--output-path demux.qza \--input-format PairedEndFastqManifestPhred33V2
##导入数据可视化:
qiime demux summarize \
> --i-data ./demux.qza \
> --o-visualization ./demux.qzv
将qzv文件在该网站中查看view.qiime2.org



生成特征表和代表序列
切除序列两端的引物,左端barcode序列(10bp)+19bp上游V3区引物,右端V4为22bp下游引物。我这里barcode序列已经被切除,所以为左端为19.
time qiime dada2 denoise-paired \--i-demultiplexed-seqs demux.qza \--p-n-threads 4 \--p-trim-left-f 19 --p-trim-left-r 22 \--p-trunc-len-f 0 --p-trunc-len-r 0 \--o-table dada2-table.qza \--o-representative-sequences dada2-rep-seqs.qza \--o-denoising-stats denoising-stats.qza
# 改名
cp dada2-table.qza table.qza
cp dada2-rep-seqs.qza rep-seqs.qza
特征表和代表序列统计
qiime feature-table summarize \--i-table table.qza \--o-visualization table.qzv \--m-sample-metadata-file metadata.txt
qiime feature-table tabulate-seqs \--i-data rep-seqs.qza \--o-visualization rep-seqs.qzv
下载qzv文件并在view.qiime2.org查看,根据特征表确定抽平深度。


Alpha和beta多样性分析
构建进化树用于多样性分析
qiime phylogeny align-to-tree-mafft-fasttree \--i-sequences rep-seqs.qza \--o-alignment aligned-rep-seqs.qza \--o-masked-alignment masked-aligned-rep-seqs.qza \--o-tree unrooted-tree.qza \--o-rooted-tree rooted-tree.qza
计算核心多样性
采样深度通常选择最小值,来自table.qzv
qiime diversity core-metrics-phylogenetic \--i-phylogeny rooted-tree.qza \--i-table table.qza \--p-sampling-depth 19954 \--m-metadata-file metadata.txt \--output-dir core-metrics-results
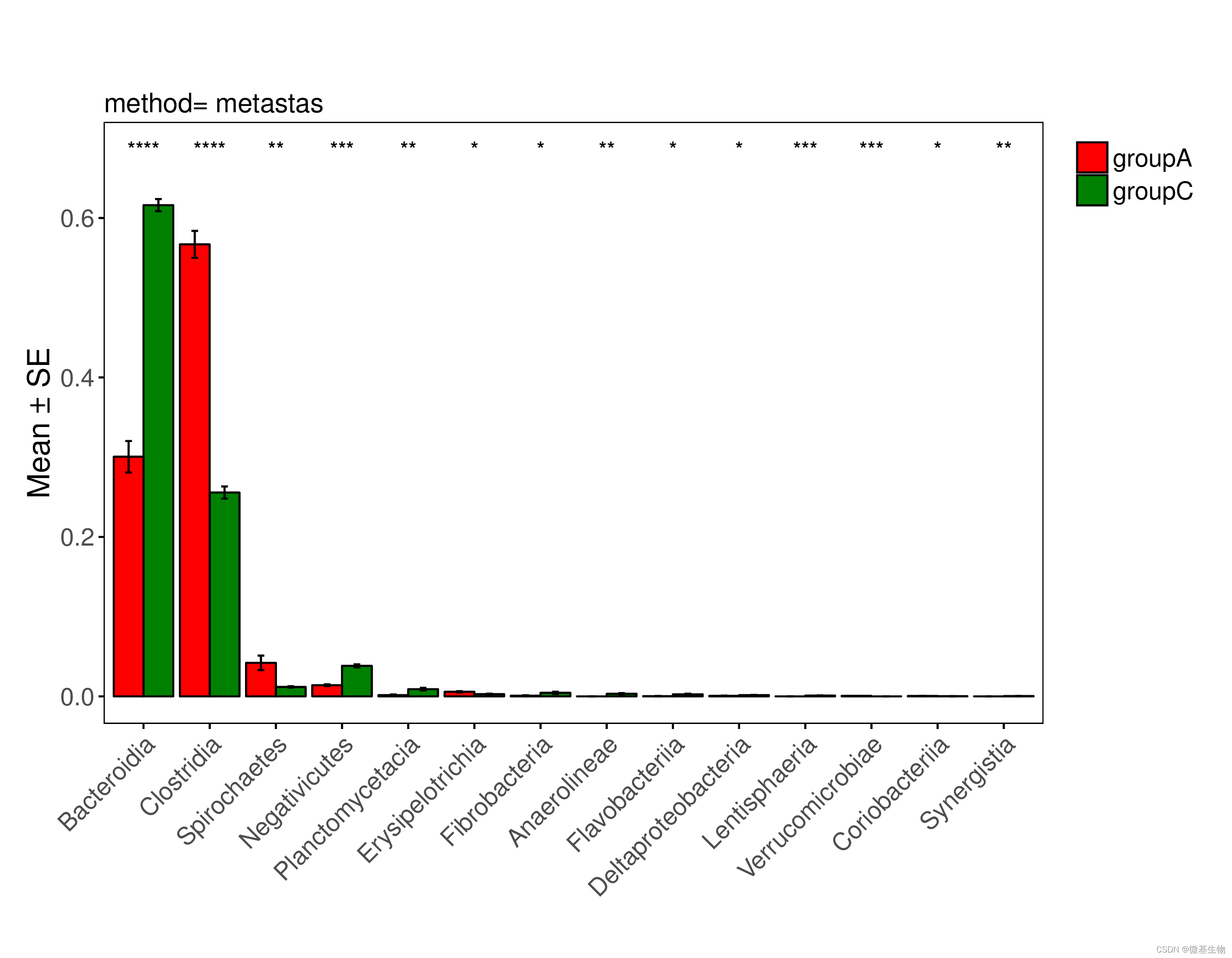
Alpha多样性组间显著性分析和可视化
可选的alpha指数有 faith_pd、shannon、observed_features、evenness
index=observed_features
qiime diversity alpha-group-significance \--i-alpha-diversity core-metrics-results/${index}_vector.qza \--m-metadata-file metadata.txt \--o-visualization core-metrics-results/${index}-group-significance.qzv
Alpha多样性稀疏曲线
max-depth选最大值,来自table.qzv
qiime diversity alpha-rarefaction \--i-table table.qza \--i-phylogeny rooted-tree.qza \--p-max-depth 78267 \--m-metadata-file metadata.txt \--o-visualization alpha-rarefaction.qzv
# 结果有observed_otus, shannon, 和faith_pd三种指数可选
Beta多样性组间显著性分析和可视化
可选的beta指数有 unweighted_unifrac、bray_curtis、weighted_unifrac和jaccard
# 7s, 指定分组是减少计算量,置换检验较耗时
distance=weighted_unifrac
column=Group
qiime diversity beta-group-significance \--i-distance-matrix core-metrics-results/${distance}_distance_matrix.qza \--m-metadata-file metadata.txt \--m-metadata-column ${column} \--o-visualization core-metrics-results/${distance}-${column}-significance.qzv \--p-pairwise
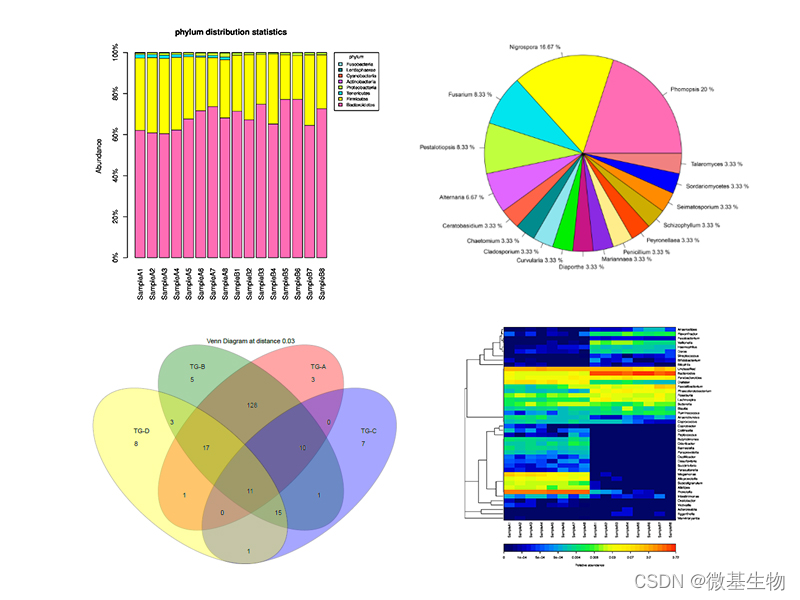
物种组成分析
物种注释,数据库见附录,可先silva-138-99-nb-classifier.qza 或 2022.10.backbone.full-length.nb.qza
1m 可选特异引物训练集如:如classifier_gg_13_8_99_V3-V4.qza 是我用V5-V7训练的文件,详见附录或官方教程
time qiime feature-classifier classify-sklearn \--i-classifier classer/silva-138-99-nb-classifier.qza \--i-reads rep-seqs.qza \--o-classification taxonomy.qza
# 可视化物种注释
qiime metadata tabulate \--m-input-file taxonomy.qza \--o-visualization taxonomy.qzv
# 堆叠柱状图展示
qiime taxa barplot \--i-table table.qza \--i-taxonomy taxonomy.qza \--m-metadata-file metadata.txt \--o-visualization taxa-bar-plots.qzv
差异分析
# 格式化特征表,添加伪计数,4s
qiime composition add-pseudocount \--i-table table.qza \--o-composition-table comp-table.qza
# 计算差异特征,指定分组类型比较,1m
column=Group
time qiime composition ancom \--i-table comp-table.qza \--m-metadata-file metadata.txt \--m-metadata-column ${column} \--o-visualization ancom-${column}.qzv# 按属水平合并,并统计
## 按属水平合并,6s
qiime taxa collapse \--i-table table.qza \--i-taxonomy taxonomy.qza \--p-level 6 \--o-collapsed-table table-l6.qza
# 格式化特征表,添加伪计数,6s
qiime composition add-pseudocount \--i-table table-l6.qza \--o-composition-table comp-table-l6.qza
# 计算差异属,指定分组类型比较,16s
qiime composition ancom \--i-table comp-table-l6.qza \--m-metadata-file metadata.txt \--m-metadata-column ${column} \--o-visualization ancom-l6-${column}.qzv
数据导出
导出科水平OTU表格
qiime taxa collapse\
> --i-table table.qza \
> --i-taxonomy taxonomy.qza \
> --p-level 5\
> --o-collapsed-table table-l5.qzaqiime tools export\
> --input-path table-l5.qza\
> --output-path exported-tablebiom convert -i exported-table/feature-table.biom\
> -o exported-table/silva_l5.txt --to-tsv
导出ASV数据
qiime tools export\
> --input-path table.qza\
> --output-path exported-table
biom convert -i exported-table/feature-table.biom\
> -o exported-table/asv_table.txt --to-tsv
物种注释数据训练集
Silva 138 99% OTUs full-length sequences
官网下载
wget -c https://data.qiime2.org/2023.5/common/silva-138-99-nb-classifier.qza
Greengenes2 2022.10 full length sequences
官网下载
wget -c ftp://download.nmdc.cn/tools/amplicon/silva/silva-138-99-nb-classifier.qza
物种注释数据训练集
下载数据库文件(greengenes, 320M)
wget -c ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz
mv gg_13_8_otus_99.tar.gz gg_13_8_otus.tar.gz
#解压
tar -zxvf gg_13_8_otus.tar.gz
使用rep_set文件中的99_otus.fasta数据和taxonomy中的99_OTU_taxonomy.txt数据作为参考物种注释
导入参考序列,50s
qiime tools import \--type 'FeatureData[Sequence]' \--input-path gg_13_8_otus/rep_set/99_otus.fasta \--output-path 99_otus.qza
导入物种分类信息,6s
qiime tools import \--type 'FeatureData[Taxonomy]' \--input-format HeaderlessTSVTaxonomyFormat \--input-path gg_13_8_otus/taxonomy/99_otu_taxonomy.txt \--output-path ref-taxonomy.qza
Train the classifier(训练分类器)——全长
time qiime feature-classifier fit-classifier-naive-bayes \--i-reference-reads 99_otus.qza \--i-reference-taxonomy ref-taxonomy.qza \--o-classifier classifier_gg_13_8_99.qza
引物提取参考序列的扩增区段 Extract reference reads
常用Greengenes 13_8 99% OTUs from 341F CCTACGGGNGGCWGCAG/805R GACTACHVGGGTATCTAATCC region of sequences(分类器描述),提供测序的引物序列,截取对应的区域进行比对,达到分类的目的。

本次使用引物341F-805R,请根据实际替换,
time qiime feature-classifier extract-reads \--i-sequences 99_otus.qza \--p-f-primer CCTACGGGNGGCWGCAG \--p-r-primer GACTACHVGGGTATCTAATCC \--o-reads ref-seqs.qza
Train the classifier(训练分类器)
基于筛选的指定区段,生成实验特异的分类器
time qiime feature-classifier fit-classifier-naive-bayes \--i-reference-reads ref-seqs.qza \--i-reference-taxonomy ref-taxonomy.qza \--o-classifier classifier_gg_13_8_99_V3-V4.qza
参考文献
Evan Bolyen, Jai Ram Rideout, Matthew R. Dillon, Nicholas A. Bokulich, Christian C. Abnet, Gabriel A. Al-Ghalith, Harriet Alexander, Eric J. Alm, Manimozhiyan Arumugam, Francesco Asnicar, Yang Bai, Jordan E. Bisanz, Kyle Bittinger, Asker Brejnrod, Colin J. Brislawn, C. Titus Brown, Benjamin J. Callahan, Andrés Mauricio Caraballo-Rodríguez, John Chase, Emily K. Cope, Ricardo Da Silva, Christian Diener, Pieter C. Dorrestein, Gavin M. Douglas, Daniel M. Durall, Claire Duvallet, Christian F. Edwardson, Madeleine Ernst, Mehrbod Estaki, Jennifer Fouquier, Julia M. Gauglitz, Sean M. Gibbons, Deanna L. Gibson, Antonio Gonzalez, Kestrel Gorlick, Jiarong Guo, Benjamin Hillmann, Susan Holmes, Hannes Holste, Curtis Huttenhower, Gavin A. Huttley, Stefan Janssen, Alan K. Jarmusch, Lingjing Jiang, Benjamin D. Kaehler, Kyo Bin Kang, Christopher R. Keefe, Paul Keim, Scott T. Kelley, Dan Knights, Irina Koester, Tomasz Kosciolek, Jorden Kreps, Morgan G. I. Langille, Joslynn Lee, Ruth Ley, Yong-Xin Liu, Erikka Loftfield, Catherine Lozupone, Massoud Maher, Clarisse Marotz, Bryan D. Martin, Daniel McDonald, Lauren J. McIver, Alexey V. Melnik, Jessica L. Metcalf, Sydney C. Morgan, Jamie T. Morton, Ahmad Turan Naimey, Jose A. Navas-Molina, Louis Felix Nothias, Stephanie B. Orchanian, Talima Pearson, Samuel L. Peoples, Daniel Petras, Mary Lai Preuss, Elmar Pruesse, Lasse Buur Rasmussen, Adam Rivers, Michael S. Robeson, Patrick Rosenthal, Nicola Segata, Michael Shaffer, Arron Shiffer, Rashmi Sinha, Se Jin Song, John R. Spear, Austin D. Swafford, Luke R. Thompson, Pedro J. Torres, Pauline Trinh, Anupriya Tripathi, Peter J. Turnbaugh, Sabah Ul-Hasan, Justin J. J. van der Hooft, Fernando Vargas, Yoshiki Vázquez-Baeza, Emily Vogtmann, Max von Hippel, William Walters, Yunhu Wan, Mingxun Wang, Jonathan Warren, Kyle C. Weber, Charles H. D. Williamson, Amy D. Willis, Zhenjiang Zech Xu, Jesse R. Zaneveld, Yilong Zhang, Qiyun Zhu, Rob Knight, J. Gregory Caporaso. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37: 852-857. https://doi.org/10.1038/s41587-019-0209-9
IF: 68.164 Q1 B1