
跟着Cell学作图 | 6.时间序列分析(Mfuzz包)
一个答疑教程。

示例数据
#BiocManager::install('maSigPro')
library(maSigPro)
# 载入示例数据
data(data.abiotic)
data.abiotic[1:5,1:5]
data(edesign.abiotic)
head(edesign.abiotic)> data.abiotic[1:5,1:5]Control_3H_1 Control_3H_2 Control_3H_3 Control_9H_1 Control_9H_2
STMDF90 0.13735714 -0.3653065 -0.15329448 0.44754535 0.287476796
STMCJ24 NA NA NA NA NA
STMJH42 0.07864449 0.1002328 -0.17365488 -0.25279484 0.184855409
STMDE66 0.22976991 0.4740975 0.46930716 0.37101059 -0.004992029
STMIX74 0.14407618 -0.4801864 -0.07847999 0.05692331 0.013045420> head(edesign.abiotic)Time Replicate Control Cold Heat Salt
Control_3H_1 3 1 1 0 0 0
Control_3H_2 3 1 1 0 0 0
Control_3H_3 3 1 1 0 0 0
Control_9H_1 9 2 1 0 0 0
Control_9H_2 9 2 1 0 0 0
Control_9H_3 9 2 1 0 0 0注意:data.abiotic是已经标准化过的基因表达矩阵。
建立回归模型
生成回归矩阵(makeDesignMatrix)
design <- make.design.matrix(edesign.abiotic, degree = 2)
design$groups.vector示例数据有三个时间的,故考虑二次回归模型(degree = 2)。
> design$groups.vector[1] "ColdvsControl" "HeatvsControl" "SaltvsControl" "Control" [5] "ColdvsControl" "HeatvsControl" "SaltvsControl" "Control" [9] "ColdvsControl" "HeatvsControl" "SaltvsControl"寻找重要基因(p.vector)
F检验确定回归方程的显著性,采用BH的校正方式,校正多重假设检验的p值。
校正后的p值小于p.vector的参数Q的基因就作为候选基因,进行下一步的分析。通过fit$SELEC可以得到候选基因的表达量信息。
fit <- p.vector(data.abiotic, # 标准化的表达矩阵 design, # 实验设计的矩阵 make.design.matrix 生成Q = 0.05, # 显著性水平MT.adjust = "BH", min.obs = 20 # 最低表达样本数 不应小于(degree+1)xGroups+1 )fit$i # 显著性基因的数量
fit$SELEC # 显著性基因表达矩阵寻找显著性差异(T.fit)
上述的回归方程是基于所有的自变量的组合构建的,接下来就是通过逐步回归法确定最佳的自变量组合。
tstep <- T.fit(fit, # p.vector结果step.method = "backward", alfa = 0.05) # 在逐步回归中用于变量选择的显著性水平在挑选最佳的自变量组合时,通过每种自变量组合对应的回归模型的拟合优度值R-squared来进行判断,R-squared取值范围为0到1,数值越大,越接近1,回归模型的效果越好。
获取显著性基因列表(get.siggenes)
sigs <- get.siggenes(tstep, # T.fit结果rsq = 0.6, # 逐步回归中的R-squared截至值vars = "groups")
# vars参数有3种
# all 每个基因直接给出一个最佳的回归模型
# groups 只给出不同实验条件下相比control组中的差异基因
# each 会给出时间点和实验条件的所有组合对应差异基因列表names(sigs)
names(sigs$sig.genes$ColdvsControl)
sigs$sig.genes$ColdvsControl$sig.profiles # 查看cold vs control的结果结果可视化
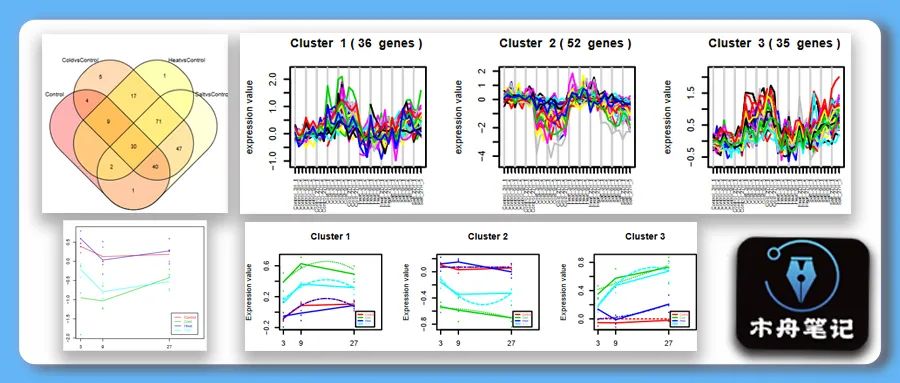
韦恩图(suma2Venn)
suma2Venn(sigs$summary[, c(2:4)]) # 左图
suma2Venn(sigs$summary[, c(1:4)]) # 右图
# 这个韦恩图面积大小与数量不成比例 较普通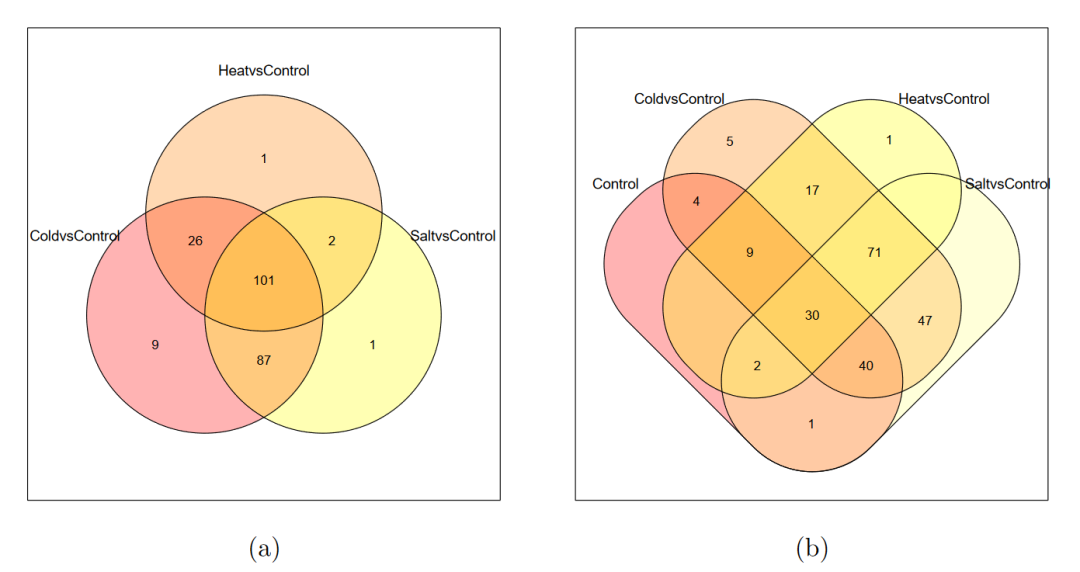
see.genes()
see.genes(sigs$sig.genes$ColdvsControl, # 差异基因表达矩阵show.fit = T, # 是否显示回归拟合线(虚线)dis =design$dis, # 回归设计矩阵cluster.method="hclust" , # 聚类方法cluster.data = 1, k = 9) # 聚类数目
这一步生成两个图,如图可分别查看。注意调整图片显示区域大小,以免报错。
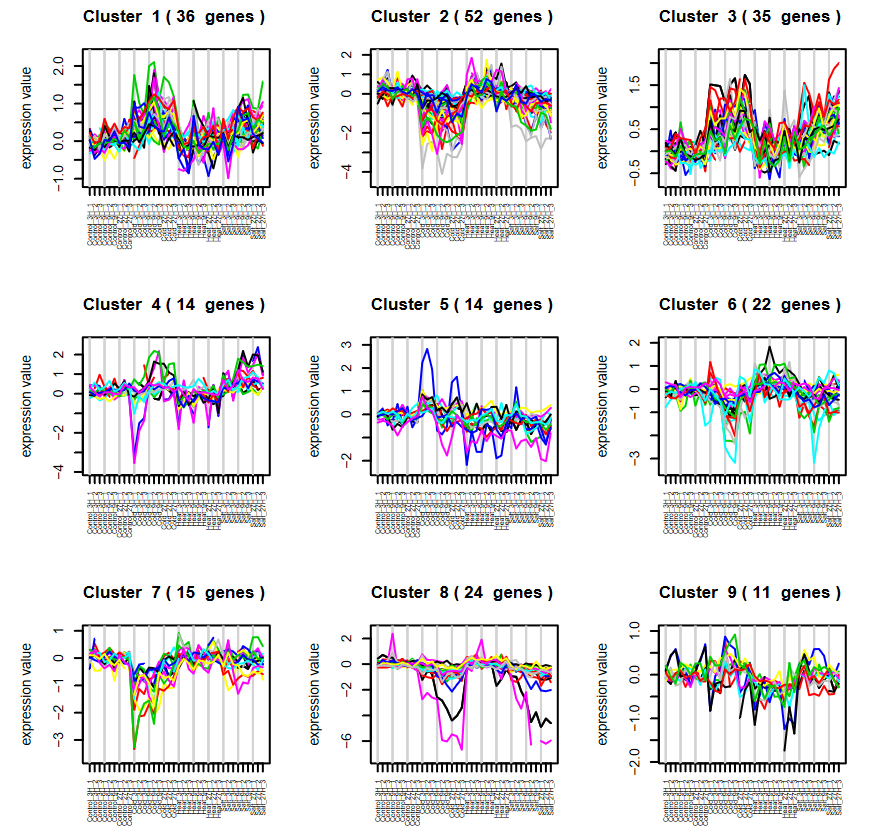
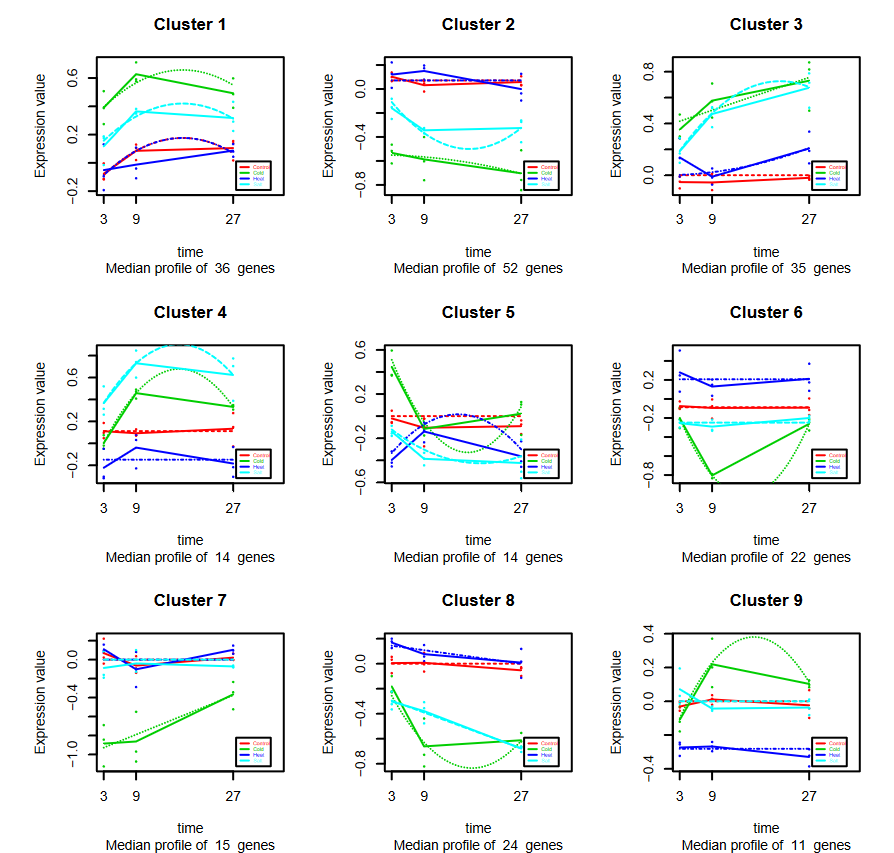
PlotGroups()
选择某一特定genes的表达进行可视化。
# 选取STMDE66基因
STMDE66 <- data.abiotic[rownames(data.abiotic)=="STMDE66", ]
PlotGroups (STMDE66, edesign = edesign.abiotic)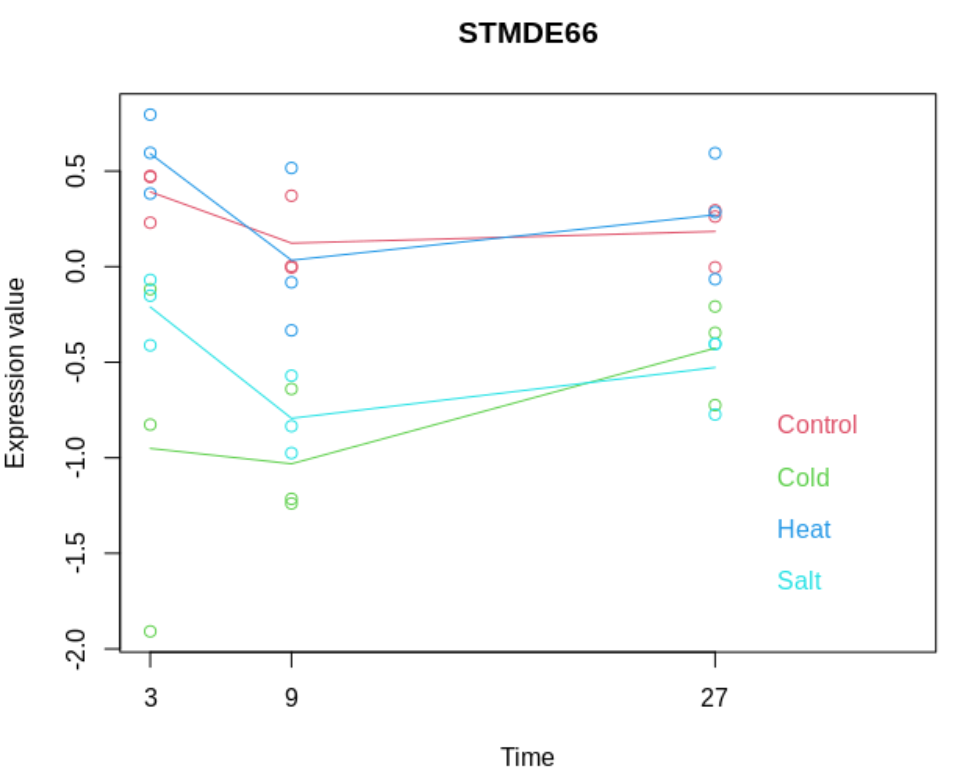
# 添加回归拟合线
PlotGroups (STMDE66, edesign = edesign.abiotic, show.fit = T, dis = design$dis, groups.vector = design$groups.vector)
示例数据和代码领取
点赞、在看 本文,分享至朋友圈集赞20个并保留30分钟,截图发至微信mzbj0002领取。
木舟笔记2022年度VIP可免费领取。
木舟笔记2022年度VIP企划
权益:
2022年度木舟笔记所有推文示例数据及代码(在VIP群里实时更新)。

木舟笔记科研交流群。
半价购买
跟着Cell学作图系列合集(免费教程+代码领取)|跟着Cell学作图系列合集。
收费:
99¥/人。可添加微信:mzbj0002 转账,或直接在文末打赏。

参考
Bioconductor - maSigPro(https://bioconductor.org/packages/release/bioc/html/maSigPro.html)
往期内容
(免费教程+代码领取)|跟着Cell学作图系列合集
Q&A | 如何在论文中画出漂亮的插图?
Front Immunol 复现 | 4. 使用estimate包评估肿瘤纯度
R绘图 | 气泡散点图+拟合曲线
跟着 Cell 学作图 | 桑葚图(ggalluvial)
R绘图 | 对比条形图+连线
R绘图 | 一幅小提琴图的美化之旅
R实战 | 给聚类加个圈圈(ggunchull)
R绘图 | 描述性统计常用图(散点图+柱状图+饼图)

![NGS数据分析实践:03. 涉及的常用数据格式[5] - vcf格式](https://img-blog.csdnimg.cn/d0d7202c70aa4f098cdb89aa94180718.png?x-oss-process=image/watermark,type_ZHJvaWRzYW5zZmFsbGJhY2s,shadow_50,text_Q1NETiBAaHVjeV9CaW9pbmZv,size_20,color_FFFFFF,t_70,g_se,x_16)



![NGS数据分析实践:05. 测序数据的基本质控 [2] - MultiQC](https://img-blog.csdnimg.cn/b50e98ac57304a7faececee2b905a970.png?x-oss-process=image/watermark,type_ZmFuZ3poZW5naGVpdGk,shadow_10,text_aHR0cHM6Ly9ibG9nLmNzZG4ubmV0L3FxXzIyMjUzOTAx,size_16,color_FFFFFF,t_70)
![NGS数据分析实践:03. 涉及的常用数据格式[2] - sam/bam格式](https://img-blog.csdnimg.cn/ab8a7d33b59c48479415397aa1fee723.png?x-oss-process=image/watermark,type_ZHJvaWRzYW5zZmFsbGJhY2s,shadow_50,text_Q1NETiBAaHVjeV9CaW9pbmZv,size_20,color_FFFFFF,t_70,g_se,x_16)