NGS数据分析实践:03. 涉及的常用数据格式[5] - vcf格式
- 6. vcf格式
- 6.1 vcf格式整体描述
- 6.2 第8列INFO详解
- 6.3 第9列FORMAT详解
- 6.4 vcf文件简单解读
系列文章:
二代测序方法:DNA测序之靶向重测序
NGS数据分析实践:00. 变异识别的基本流程
NGS数据分析实践:01. Conda环境配置及软件安装
NGS数据分析实践:02. 参考基因组及注释库的下载
NGS数据分析实践:03. 涉及的常用数据格式[1] - fasta和fastq格式
NGS数据分析实践:03. 涉及的常用数据格式[2] - sam/bam格式
NGS数据分析实践:03. 涉及的常用数据格式[3] - gtf/gff格式
NGS数据分析实践:03. 涉及的常用数据格式[4] - bed和Wiggle/Bigwig/bedgraph格式
测序数据分析中涉及的常用格式:测序得到的是带有质量值的碱基序列(fastq格式),参考基因组是(fasta格式),用比对工具把fastq格式的序列比对到对应的fasta格式的参考基因序列,就可以产生sam格式的比对文件。把sam格式的文本文件压缩成二进制bam文件可以节省空间,如果对参考基因组上面的各个区段标记它们的性质,比如哪些区域是外显子、内含子、UTR等等,这就是gtf/gff格式。如果只是为了单纯描述某个基因组区域,就是bed格式文件,记录染色体号以及起始终止坐标,正负链即可。如果是记录某些位点或者区域碱基的变异,就是vcf文件格式。
fasta/fastq(测序数据)→SAM/BAM(比对)→gff/gtf(描述基因组上的结构:坐标&类型)→Bigwig/Wiggle(测序深度)→bed(描述坐标)→vcf(突变信息)
存储序列:fasta/fastq
比对结果显示的文件:sam/bam
展示注释信息:gtf/gff/bed
突变信息:vcf
6. vcf格式
VCF格式全称为 Variant Call Format,是存储变异位点的标准格式,可以用来表示单核苷酸多态性(SNP)【在人类基因组中,分布普遍并且密度比较大,总数超过107, 平均每300bp就有一个SNP 】、插入缺失(InDel,也就是短片段的插入与缺失)、结构变异(SV:Structural Variant,也就是大片段的插入与缺失) 、拷贝数量变异(CNV:Copy Number Variant)【CNV:比如一个基因在染色体的一条染色单体上的数目为1,但是在染色体复制过程中,不知为何,复制结束后该基因在染色单体数目由1变成了2或者n。尤其在人类基因组中存在大量大于1 kb但小于3 Mb的DNA片段多态。它发生的频率远远高于染色体结构变异,并且整个基因组中覆盖的核苷酸总数大大超过SNP的总数】。VCF格式在GATK软件中得到很好的支持。
官方说明详见:http://gatkforums.broadinstitute.org/discussion/1268/how-should-i-interpret-vcf-files-produced-by-the-gatk
6.1 vcf格式整体描述
VCF文件分为两部分内容:以“##”开头的meta信息部分(key=value)和主体数据部分;可理解为注释部分 + 主体部分。
注释部分有很多对VCF的介绍信息;主体部分每一行代表一个variant的信息。单样本的vcf文件一般含10列数据:CHROM、POS、ID、REF、ALT、QUAL、FILTER、INFO、FORAMT、SAMPLE。其中,前8列为固定的必须要有的列,各列以制表符(\t)分割。

以下为每一列的说明:

注意:FORMAT & SAMPLE:两部分共同描述了变异位点的基本统计信息,不同信息之间由“:”分隔。每一个sample对应着1列;多个samples则对应着多列,这种情况下,列的数多余10列,且个sample列的ID不能重复。各标签代表的意义在注释部分##FORMAT有详细说明。
例如:FORMAT为GT:AD:DP:GQ:PL,SAMPLE为0/1:16,11:27:99:251,0,375,表示该样本的该变异位点GT(Genotype)为0/1,AD(Allele Depth)为16,11,DP(Read Depth)为27,GQ(Genotype Quality)为99,PL(provides the likelihoods of the given genotypes)为251,0,375。
注:不同的变异识别软件生成的vcf文件中,INFO和FORAMT两列的tags可能略有不同,含义可参照注释部分,或者软件使用说明。
6.2 第8列INFO详解
第8列(INFO)的信息包括18种,都是以“TAG=Value”,并使用分号分隔的形式,其中很多的注释信息在VCF文件的头部注释中给出,下面对常用的TAG进行解释:
和等位基因有关的3个TAGs:
AN(Allele Number): alleles的总数目。
AC(Allele Count): 基因型为variant allele的数目。
AF(Allele Frequency): variant allele的频率,AF值=AC值/AN值。
例子1:对2个sample的二倍体进行测序,则AN值为4。若REF上位点碱基为A,2个sample在该位点分别为A/T和T/G;则AC=1;AF=0.25(在该位点只有25%的等位基因发生突变)。
例子2:对1个sample的二倍体测序,则AN=2。若基因型为杂合突变0/1,则该位点只有一个等位基因发生突变,AC=1,AF=0.5;若基因型为纯合突变1/1,AC=2,AF=1。
DP(reads覆盖度):一部分reads被过滤掉后的覆盖度。
DP4 : 高质量测序碱基,包含4个值:ref-forward , ref-reverse, alt-forward, alt-reverse。
Dels:Fraction of reads containing spanning delections,这个值用来区分indel和snv,有这个tag且为0时表示该位点是SNV,没有就是InDel。【一个物种中该单碱基变异的频率达到一定水平就叫SNP,而频率很低或未知就叫SNV】
FS:FisherStrand的缩写,表示使用Fisher’s精确检验来检测strand bias而得到的Fhred格式的p值,该值越小越好;如果该值较大,表示strand bias(正负链偏移)越严重,即所检测到的variants位点上,reads比对到正负义链上的比例不均衡。一般进行filter的时候,推荐保留FS<10~20的variants位点。GATK可设定FS参数。
ReadPosRandSum:Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias.当variants出现在reads尾部的时候,其结果可能不准确。该值用于衡量alternative allele(变异的等位基因)相比于reference allele(参考基因组等位基因),其variant位点是否匹配到reads更靠中部的位置。因此只有基因型是杂合且有一个allele和参考基因组一致的时候,才能计算该值。若该值为正值,表明和alternative allele相当于reference allele,落来reads更靠中部的位置;若该值是负值,则表示alternative allele相比于reference allele落在reads更靠尾部的位置。 进行filter的之后,推荐保留ReadPosRankSum>-1.65~-3.0的variant位点
MQRankSum: 该值用于衡量alternative allele上reads的mapping quality与reference allele上reads的mapping quality的差异。若该值是负数值,则表明alternative allele比reference allele的reads mapping quality差。进行filter的时候,推荐保留MQRankSum>-1.65~-3.0的variant位点。
6.3 第9列FORMAT详解
VCF文件主体部分的第9列是基因型信息的多个标签,这些标签之间以冒号分割,其对应的值位于第10列,同样以冒号分割,表示第一个样本的基因型结果。若有多个样本,则VCF文件超过10列,且第10列后的每一列表示一个样品的基因型结果。各标签的具体含义,需参照该文件的标头注释。
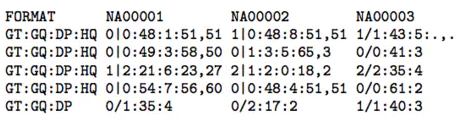
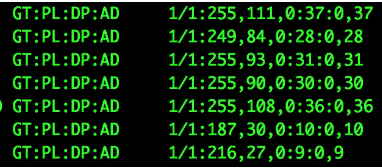
第9列(FORMAT) 各个标签的意义展示如下:
GT:genotype
样本(sample)的基因型(genotype),两个数字中间用‘/’分开,这两个数字表示双倍体的sample的基因型。0表示样品中有reference的allele(可初步理解为和ref的碱基相同,即和REF列相同);1表示样本中的variant的allele(可以理解为和variant变异后的碱基相同,即和ALT列相同);2表示有第二个variant的allele(和ALT列的第二种碱基相同)对于SNP是指单个碱基类型相同而对于Indel是指碱基类型及个数均相同。
因此,根据GT的结果得出以下结论:
0/0表示sample中该位点为纯合位点,和REF的碱基类型一致;
0/1表示sample中该位点为杂合突变,有REF和ALT两个基因型(部分碱基和REF碱基类型一致,部分碱基和ALT碱基类型一致);
1/1表示sample中该位点为纯合突变,总体突变类型和ALT碱基类型一致;
1/2表示sample中该位点为杂合突变,有ALT1和ALT2两个基因型(部分和ALT1碱基类型一致,部分和ALT2碱基类型一致)。
AD和DP
AD(Allele Depth) 为sample中每一种allele(等位碱基)的reads覆盖度,在diploid(二倍体,或可指代多倍型)中则是用逗号分隔的两个值,前者对应REF基因,后者对应ALT基因型;
DP(Depth) 为sample中该位点的覆盖度,是所支持的两个AD值(逗号前和逗号后)的加和。【第8列也有DP,但含义不同】
例如:
GT:AD(REF),AD(ALT):DP
1/1:0,175:175
0/1:79,96:175
1/2:0,20,56:76
这里的三种类型对应的DP值均是其对应的AD值的加和,1/1的175是0+175,0/1的175是79+96,1/2的76是0+20+56。
GQ(Genotype Quality)
基因型的质量值。Phred格式(Phred_scaled)的质量值,表示在该位点该基因型存在的可能性;该值越高,则Genotype的可能性越大;计算方法:Phred值=-10*log(1-P),P为基因型存在的概率。(一般在final.snp.vcf文件中,该值为99,为99时,其可能性最大)。
PL(likelihood genotypes)
指定的三种基因型的质量值(provieds the likelihoods of the given genotypes);这三种指定的基因型为(0/0,0/1,1/1),这三种基因型的概率总和为1。该值越大,表明为该种基因型的可能性越小。Phred值=-10*log§,P为基因型存在的概率。最有可能的genotype的值为0。
例如:
0/0:0,889,216 (0/0型3个数字,第一个为0);
0/1:94,0,940 (0/1型3个数字,中间为0);
1/1:269,18,0 (1/1型3个数字,最后一个为0);
1/2:3365,1522,1357,1842,0,1706 (0/0,0/1,1/1,0/2,1/2,2/2 ?)(1/2型6个数字,倒数第二个为0)。
更多信息见官方说明文档:https://samtools.github.io/hts-specs/VCFv4.2.pdf
6.4 vcf文件简单解读
用VCFv4.2官方文件中的例子,做一个简单的展示:

① 主体部分第1行:一个高质量的简单SNP,碱基质量值为29;
② 主体部分第2行:一个可能会被过滤掉的SNP,因为质量值<10;
③ 主体部分第3行:第4列REF等位为A,call出了两个alternate alleles,其中一个为T。INFO列中显示AA=T,表明T是祖先等位,因此,该变异可能是参考测序错误;
④ 主体部分第4行:该位点是单态性SNP,没有其他等位。
⑤ 主体部分第5行:包含两个alternate alleles的微卫星,即2个碱基的删除(TC)和1个碱基的插入(T)。
示例中包含3个样本,FORMAT未TAG,sample列为相应的取值value,包括:基因型(GT)、每个样本的碱基质量(GQ)、深度(DP)和单体型质量(HQ)等。
其他SNP和INDELs的示例:



参考阅读:
http://genome.ucsc.edu/FAQ/FAQformat.html
VCF格式:https://www.jianshu.com/p/957efb50108f
生信宝典 生信分析过程中这些常见文件的格式以及查看方式你都知道吗?
常用生物信息学格式介绍:http://ju.outofmemory.cn/entry/193943
https://samtools.github.io/hts-specs/VCFv4.2.pdf
https://samtools.github.io/hts-specs/VCFv4.3.pdf



![NGS数据分析实践:05. 测序数据的基本质控 [2] - MultiQC](https://img-blog.csdnimg.cn/b50e98ac57304a7faececee2b905a970.png?x-oss-process=image/watermark,type_ZmFuZ3poZW5naGVpdGk,shadow_10,text_aHR0cHM6Ly9ibG9nLmNzZG4ubmV0L3FxXzIyMjUzOTAx,size_16,color_FFFFFF,t_70)
![NGS数据分析实践:03. 涉及的常用数据格式[2] - sam/bam格式](https://img-blog.csdnimg.cn/ab8a7d33b59c48479415397aa1fee723.png?x-oss-process=image/watermark,type_ZHJvaWRzYW5zZmFsbGJhY2s,shadow_50,text_Q1NETiBAaHVjeV9CaW9pbmZv,size_20,color_FFFFFF,t_70,g_se,x_16)