步骤
- 0. 练习前准备
- 1. 找到文章对应的数据集
- 2. 下载数据集
- 3. 与参考基因组进行比对
- 4. reads计数
- 5. 踩过的一点小坑
写在前面——之前使用的数据是单端测序,但是现在的数据基本都是双端测序。所以又找了个双端测序的例子来练习。之前在单端测序数据中,因为参考基因组注释文件找的不对,所以reads计数没有做好。这次数据质量不错,所以省略了质控和清洗,直接进入主题。由于租的服务器是2核+8G的,所以在生成sam文件和sort以及htseq-count都花费了大量的时间(四个样本集整整跑了将近一整天)。不过最后结果算是复现出来了,甚是欣慰。
文章名:AKAP95 regulates splicing through scaffolding RNAs and RNA processing factors
参考:https://www.jianshu.com/p/6d4cba26bb60
0. 练习前准备
a. 建好相关文件夹
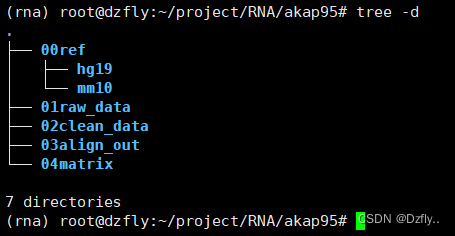
b. 00ref:存放参考基因组和基因组注释文件(红色框框内为本文需要的文件)
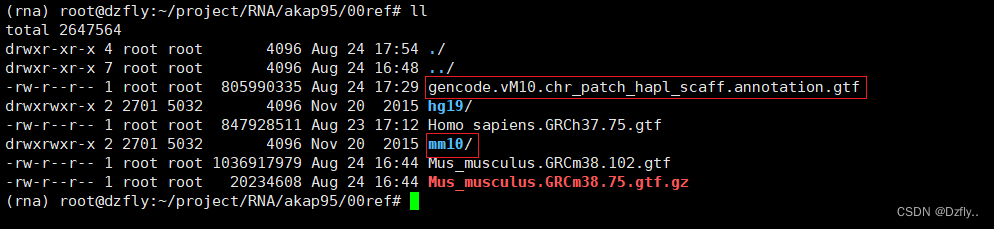
c. 01raw_data:双端测序,所以一个样本有两个文件。
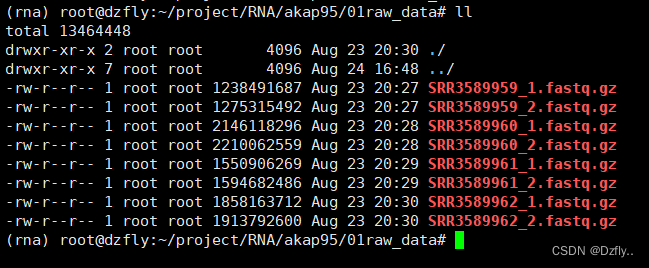
d. clean_data:存放处理过后的数据,本文数据质量不错,所以不用清洗即可使用
e. align_data:存放比对后的文件
f. matrix:存放reads计数文件
1. 找到文章对应的数据集

https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE81916
2. 下载数据集
具体:快速下载SRA数据
for ((i=59;i<=62;i++)) ;do prefetch -v SRR35899$i; done
fastq-dump --gzip --split-3 SRR35899*.sra# 在另一个Linux用户下载的,需要传到自己的目录下
scp SRR35899*.gz root@dzfly:/root/project/RNA/akap95/01raw_data/
检测公司给的数据是否完整:md5sum -c md5.txt

3. 与参考基因组进行比对
# 使用hisat2进行比对
# -t 显示时间
# -p 设置线程
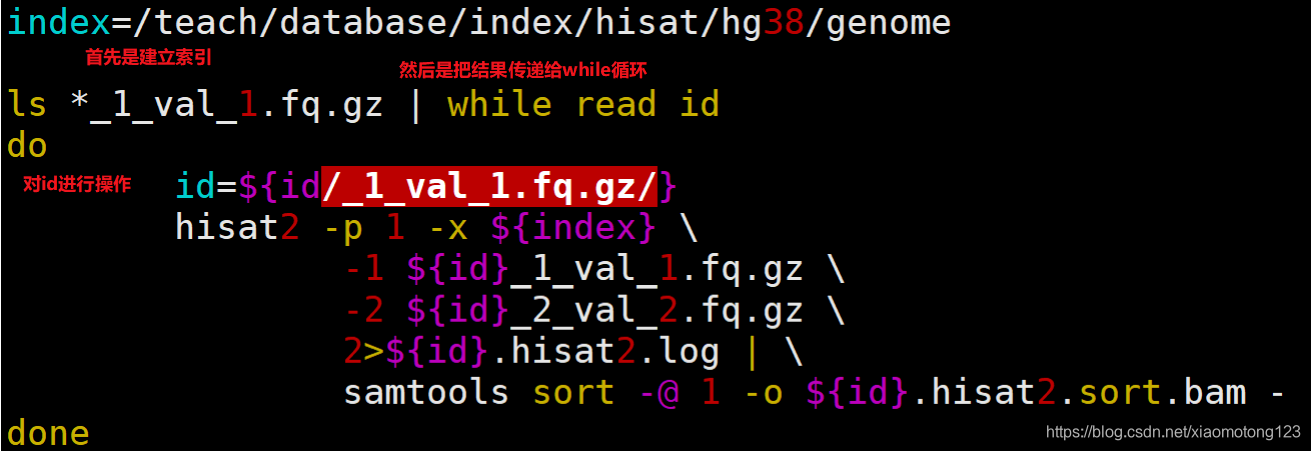
for ((i=59;i<=62;i++)); do hisat2 -t -p 2 -x 00ref/mm10/genome -1 01raw_data/SRR35899${i}_1.fastq.gz -2 01raw_data/SRR35899${i}_2.fastq.gz -S 03align_out/SRR35899${i}.sam; done# 默认为pos排序,即默认按染色体顺序排列
for ((i=59;i<=62;i++)); do samtools sort -O bam -@ 2 -o SRR35899${i}.bam SRR35899${i}.sam; done
for ((i=59;i<=62;i++)); do samtools index SRR35899${i}.bam; done# flagstat检查一下结果
for ((i=59;i<=62;i++)); do samtools flagstat -@ 2 SRR35899${i}.bam > SRR35899${i}.flagstat; done
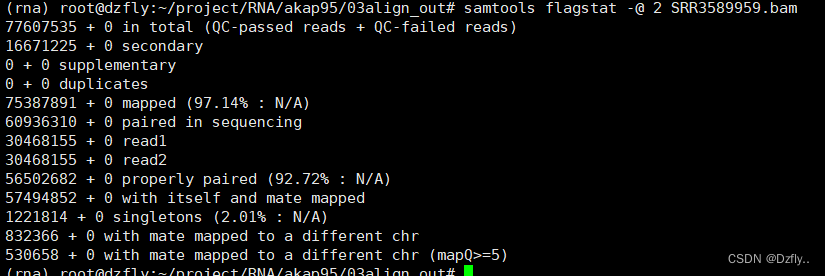
具体意义例子 参考:https://cloud.tencent.com/developer/article/1703017
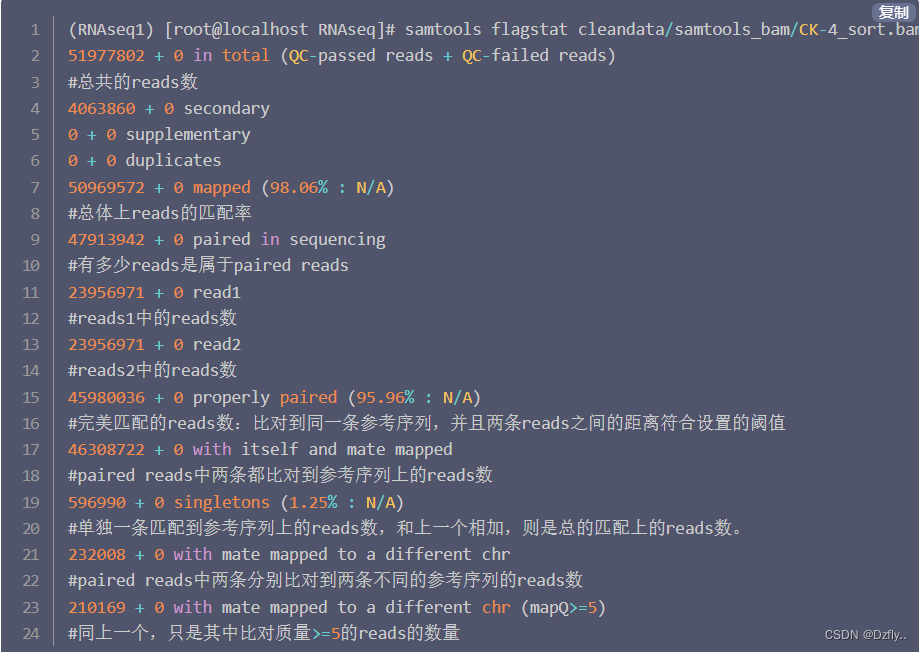
注意:sam文件真的很大,一个大概有30G。如果你的服务器磁盘容量不够,建议生成一个sam就将其转化为bam文件,然后将sam文件删除,再进行下一个。
for ((i=59;i<=62;i++)); do hisat2 -t -p 2 -x 00ref/mm10/genome -1 01raw_data/SRR35899${i}._1.fastq.gz -2 01raw_data/SRR35899${i}._2.fastq.gz -S 03align_out/SRR35899${i}.sam | samtools sort -O bam -@ 2 -o SRR35899${i}.bam SRR35899${i}.sam | samtools index SRR35899${i}.bam | rm SRR35899${i}.sam; done
4. reads计数
# 方法一
# 首先将bam文件按照name进行排序,默认为pos排序
for ((i=59;i<=62;i++)); do samtools sort -@ 2 -n SRR35899${i}.bam -o SRR35899${i}_nsorted.bam; done
# 使用htseq-count进行计算
for ((i=59;i<=62;i++)); do htseq-count -r name -f bam 03align_out/SRR35899${i}_nsorted.bam 00ref/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf > 04matrix/SRR35899${i}.count; done# 方法二
# 不按name排序,直接计算
for ((i=59;i<=62;i++)); do htseq-count -r pos -f bam 03align_out/SRR35899${i}.bam 00ref/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf > 04matrix/SRR35899${i}.count; donehead -n 4 SRR35899*.count
tail -n 4 SRR35899*.count
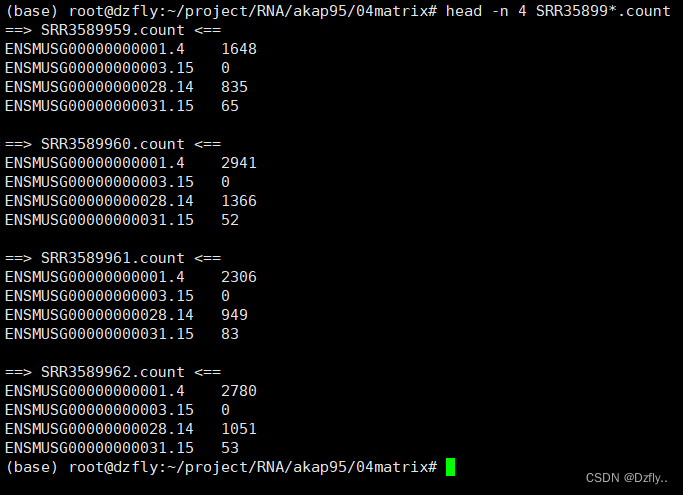
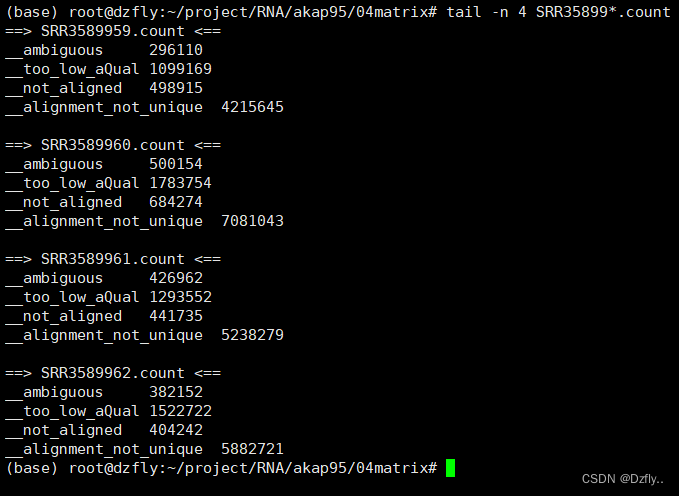
no_feature:比对区域与任何基因都没有重叠 。
ambiguous:比对区域与多个基因都发生重叠
too_low_aQual:比对质量低于设定阈值(默认是10)
not_aligned:无法比对上
alignment_not_unique:比对位置不唯一
至此,上游分析就结束了。最后获得的count文件真是来之不易呀。之后就可以使用R语言等进行分析、绘图等等了。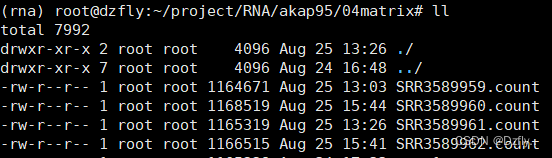
5. 踩过的一点小坑
没有找到对应的参考基因组注释文件,导致我的htseq-count结果全都是0。后来发现参数使用的也不对。具体如下:
注意 -r name;这里我用的文件其实是按默认pos排序的bam文件,所以结果都是0!!!
htseq-count -r name -f bam 03align_out/SRR3589959.bam 00ref/Mus_musculus.GRCm38.102.gtf > 04matrix/test1.count
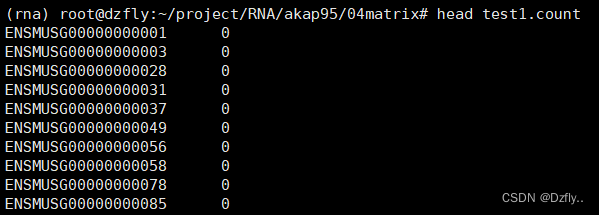
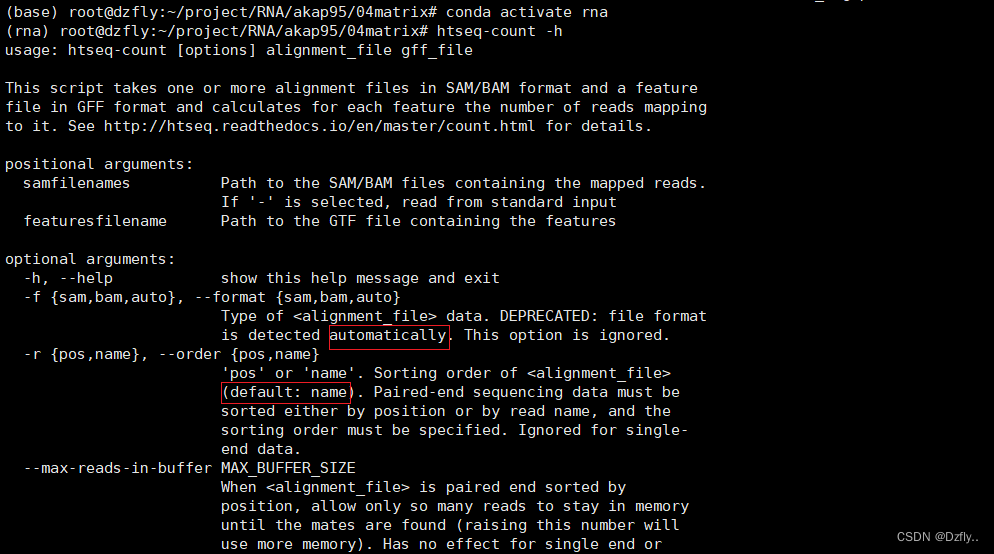
看一下htseq-count的具体参数 :
-f 可以自动识别文件类型
-r 默认处理按name排序的文件
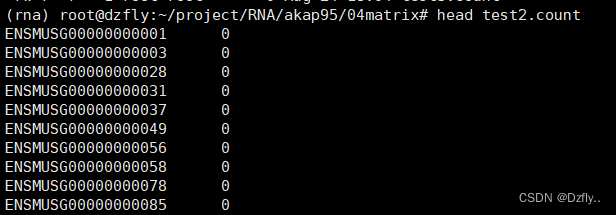
这里使用 -r pos,但是结果还是0。所以是参考基因组注释文件的问题。
htseq-count -r pos -f bam 03align_out/SRR3589959.bam 00ref/Mus_musculus.GRCm38.102.gtf > 04matrix/test2.count
找到准确的参考基因组注释文件:https://www.gencodegenes.org/mouse/release_M10.html
将文件按照name排序,再进行计算,结果终于不是0了!!!
samtools sort -n SRR3589959.bam -o SRR3589959_nsorted.bamhtseq-count -r name -f bam 03align_out/SRR3589959_nsorted.bam 00ref/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf > 04matrix/test3.count
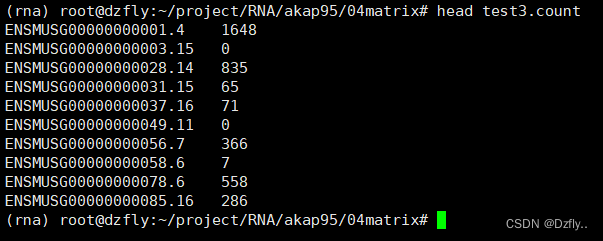
这里设置 -r pos,直接对没有按name排序的文件进行计算,可以看到结果跟test3一样。( •̀ ω •́ )y
htseq-count -n 10 -r pos -f bam 03align_out/SRR3589959.bam 00ref/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf > 04matrix/test4.count
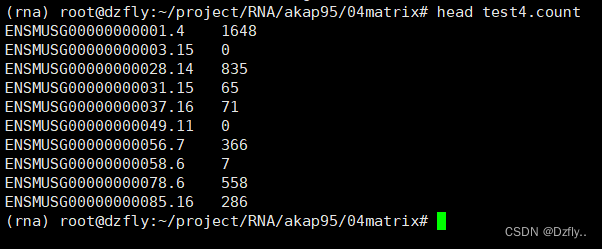
这个问题告诉我们,在处理数据之前一定要确保参考基因组和注释文件是正确的。不然会浪费很多时间!!!



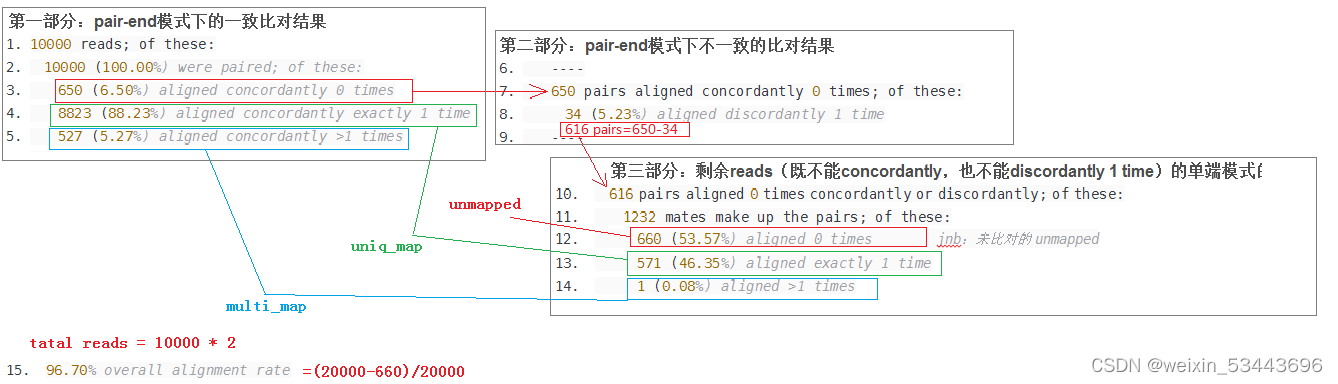
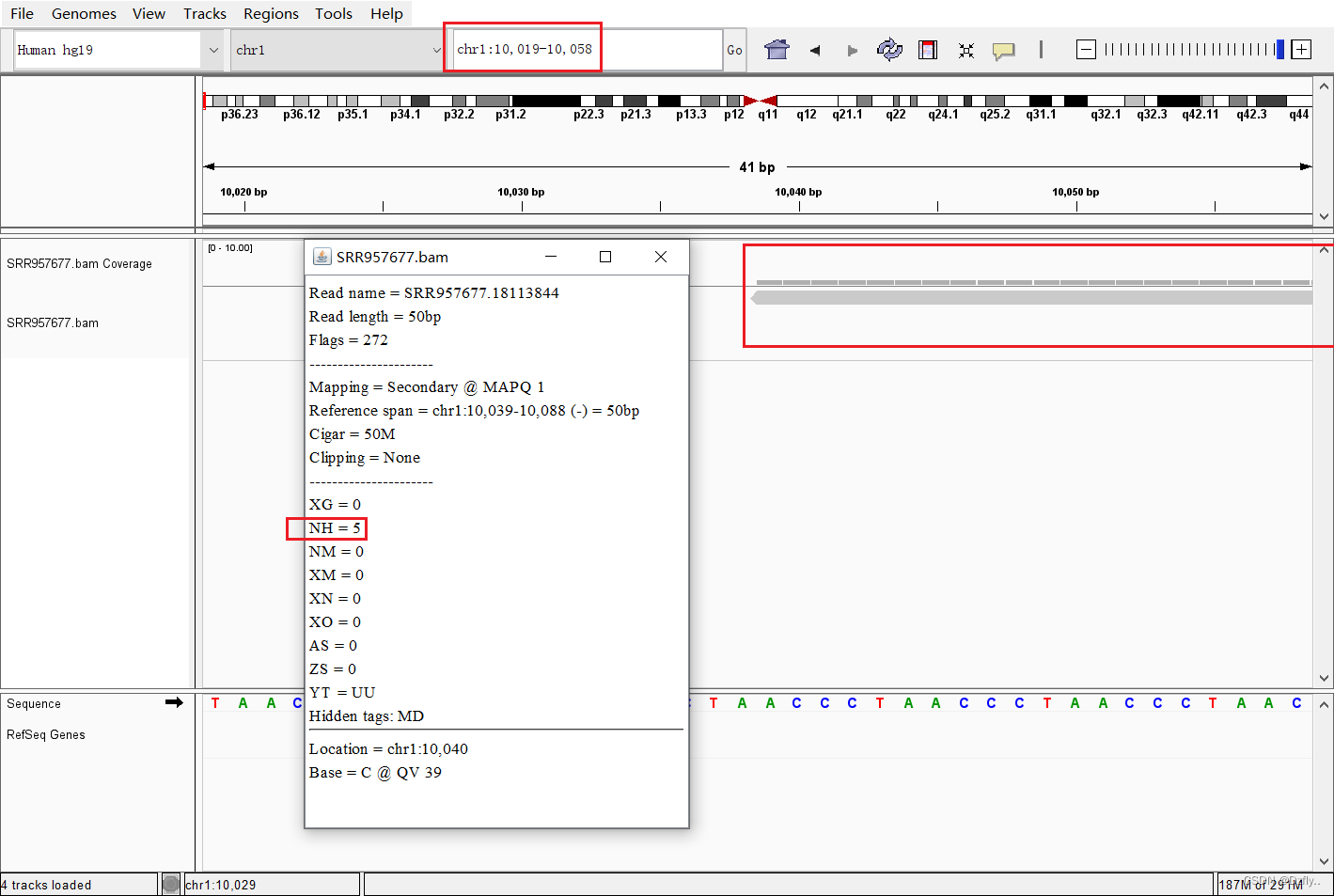


![转录组学习之序列比对(Hisat2)[学习笔记通俗易懂版]](https://img-blog.csdnimg.cn/e99c9df3c6ca436491401c75aafe334e.png)