hisat2的官网的manual:
https://github.com/DaehwanKimLab/hisat2/blob/master/MANUAL
在这里记载了详细用法和介绍,此处仅为学习笔记,和实例记录。
HISAT2 is a fast and sensitive alignment program for mapping next-generation sequencing reads (whole-genome, transcriptome, and exome sequencing data) against the general human population (as well as against a single reference genome). Based on [GCSA] (an extension of [BWT] for a graph), we designed and implemented a graph FM index (GFM),
an original approach and its first implementation to the best of our knowledge.
In addition to using one global GFM index that represents general population,
HISAT2 uses a large set of small GFM indexes that collectively cover the whole genome
(each index representing a genomic region of 56 Kbp, with 55,000 indexes needed to cover human population).
These small indexes (called local indexes) combined with several alignment strategies enable effective alignment of sequencing reads.
This new indexing scheme is called Hierarchical Graph FM index (HGFM).
We have developed HISAT 2 based on the [HISAT] and [Bowtie2] implementations.
HISAT2 outputs alignments in [SAM] format, enabling interoperation with a large number of other tools (e.g. [SAMtools], [GATK]) that use SAM.
HISAT2 is distributed under the [GPLv3 license], and it runs on the command line under
Linux, Mac OS X and Windows.
- 适用范围
RNA-SEQ
- 使用方法
1.下载数据
在hisat官网下载UCSC的数据:http://daehwankimlab.github.io/hisat2/download/

wget -c -t 0 https://genome-idx.s3.amazonaws.com/hisat/mm10_genome.tar.gz
解压缩
tar -zxvf mm10_genome.tar.gz
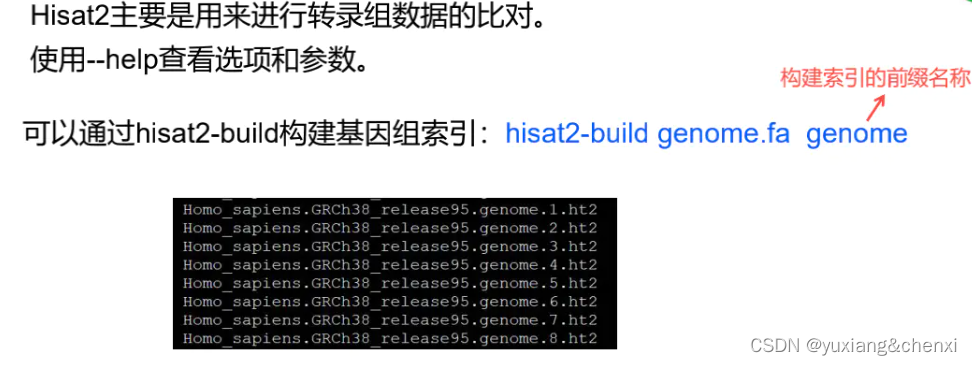
2.建立索引
hisat2-build [options]* <reference_in> <ht2_base>
$HISAT2_HOME/hisat2-build $HISAT2_HOME/example/reference/22_20-21M.fa --snp $HISAT2_HOME/example/reference/22_20-21M.snp 22_20-21M_snp
#<reference_in> :fasta文件 list,如果为list,使用逗号分开
#<ht2_base> :索引文件的前缀名,如设为xxx,则生成的索引文件为xxx.1.ht2,xxx.2.ht2,默认的前缀名为NAME
snp文件可以从官网下载
Use `hisat2_extract_snps_haplotypes_UCSC.py` (in the HISAT2 package) to extract SNPs and haplotypes from a dbSNP file (e.g. http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/snp144Common.txt.gz).
or `hisat2_extract_snps_haplotypes_VCF.py` to extract SNPs and haplotypes from a VCF file (e.g. ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/GRCh38_positions/ALL.chr22.phase3_shapeit2_mvncall_integrated_v3plus_nounphased.rsID.genotypes.GRCh38_dbSNP_no_SVs.vcf.gz).
实例:
(1)生成snp,先-h看看里面有啥要求
(riboseq) [med-zhouh@login01 hisat2]$ hisat2_extract_snps_haplotypes_UCSC.py -h
usage: hisat2_extract_snps_haplotypes_UCSC.py [-h] [--inter-gap INTER_GAP] [--intra-gap INTRA_GAP] [-v] [--testset][genome_file] [snp_fname] [base_fname]Extract SNPs and haplotypes from a SNP file downloaded from UCSC (e.g. http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/snp144.txt.gz)positional arguments:genome_file input genome file (e.g. genome.fa)snp_fname input snp file downloaded from UCSC (plain text or gzipped file is accepted: snp144Common.txt or snp144Common.txt.gz)base_fname base filename for SNPs and haplotypesoptional arguments:-h, --help show this help message and exit--inter-gap INTER_GAPMaximum distance for variants to be in the same haplotype--intra-gap INTRA_GAPBreak a haplotype into several haplotypes-v, --verbose also print some statistics to stderr--testset print test reads
看完要求就写一下,生成snp
从genecode里面下载的fasta文件。
hisat2_extract_snps_haplotypes_UCSC.py /data/med-zhouh/index/bowtie2_human_h38_index/GRCh38.p13.genome.fa snp144Common.txt h38_snp
得到结果如下:
-rw-r--r-- 1 med-zhouh med-chenh 594M Jul 28 12:33 h38_snp.haplotype
-rw-r--r-- 1 med-zhouh med-chenh 482M Jul 28 12:33 h38_snp.snp
-rw-r--r-- 1 med-zhouh med-chenh 4.8G Jan 13 2016 snp144Common.txt
(2)用刚生成的snp,和来自genecode的fa,生成索引
hisat2-build /data/med-zhouh/index/bowtie2_human_h38_index/GRCh38.p13.genome.fa --snp h38_snp hisat2_hg38_p13
得到文件如下:
-rw-r--r-- 1 med-zhouh med-chenh 992M Jul 28 14:57 hisat2_hg38_p13.1.ht2
-rw-r--r-- 1 med-zhouh med-chenh 741M Jul 28 14:57 hisat2_hg38_p13.2.ht2
-rw-r--r-- 1 med-zhouh med-chenh 17K Jul 28 14:14 hisat2_hg38_p13.3.ht2
-rw-r--r-- 1 med-zhouh med-chenh 741M Jul 28 14:14 hisat2_hg38_p13.4.ht2
-rw-r--r-- 1 med-zhouh med-chenh 1.3G Jul 28 15:05 hisat2_hg38_p13.5.ht2
-rw-r--r-- 1 med-zhouh med-chenh 754M Jul 28 15:05 hisat2_hg38_p13.6.ht2
-rw-r--r-- 1 med-zhouh med-chenh 12 Jul 28 14:14 hisat2_hg38_p13.7.ht2
-rw-r--r-- 1 med-zhouh med-chenh 8 Jul 28 14:14 hisat2_hg38_p13.8.ht2
-rw-r--r-- 1 med-zhouh med-chenh 41G Jul 28 13:16 hisat2_hg38_p13.rf
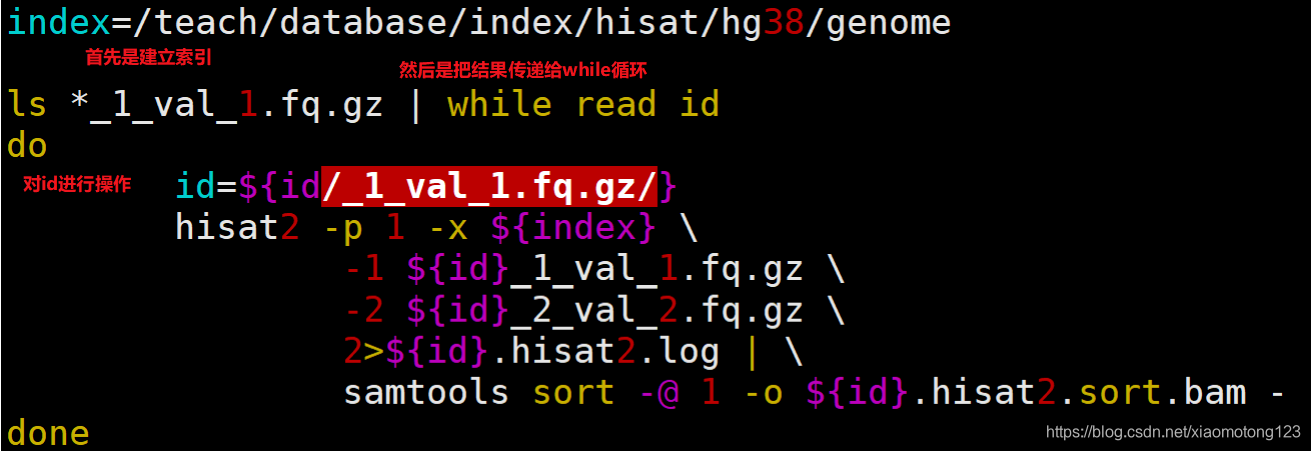
2.比对
官网实例:
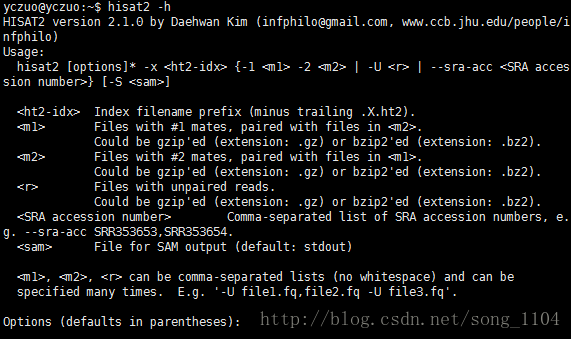
hisat2 [options]* -x <hisat2-idx> {-1 <m1> -2 <m2> | -U <r> | --sra-acc <SRA accession number>} [-S <hit>]$HISAT2_HOME/hisat2 -f -x $HISAT2_HOME/example/index/22_20-21M_snp -U $HISAT2_HOME/example/reads/reads_1.fa -S eg1.sam#-p :线程数目
#--dta :注意!!!在下游使用stringtie组装的时候一定要在hisat中设置这个参数!!!
#-x <hisat2-idx> :参考基因组索引的basename,即前缀名
#{}:其中的内容意思为hisat2可以接受单端测序,双端测序,或者直接提交SRA ID号
#-1 <m1> :双端测序的read1 list ,若为list,使用逗号隔开,名字与2要匹配,如-1 flyA_1.fq,flyB_1.fq
#-2 <m2> :双端测序的read2 list ,若为list,使用逗号隔开,名字与1要匹配,如-2 flyA_2.fq,flyB_2.fq
#-U <r>:单端测序list,若为list,使用逗号隔开,-U lane1.fq,lane2.fq,lane3.fq,lane4.fq
#--sra-acc <SRA accession number> : SRAID list,若为list,使用逗号隔开,--sra-acc SRR353653,SRR353654
#-S <hit> :SAM写入的文件名,默认写入到标准输出中
单末端
##官网例子 :$HISAT2_HOME/hisat2 -f -x $HISAT2_HOME/example/index/22_20-21M_snp -U $HISAT2_HOME/example/reads/reads_1.fa -S eg1.sam
hisat2 -f -x /xx/mm10/genome -U/xx/SRR12207279_trimmed.fq -S /xx/xx/SRR12207279.sam
双末端
##官网例子:$HISAT2_HOME/hisat2 -f -x $HISAT2_HOME/example/index/22_20-21M_snp -1 $HISAT2_HOME/example/reads/reads_1.fa -2 $HISAT2_HOME/example/reads/reads_2.fa -S eg2.sam
hisat2 -f -x /xx/mm10/genome -1 /xx/SRR12207279_1_trimmed.fq -2 /xx/xx/SRR12207279_2_trimmed.fq -S /xx/xx/SRR12207279.sam
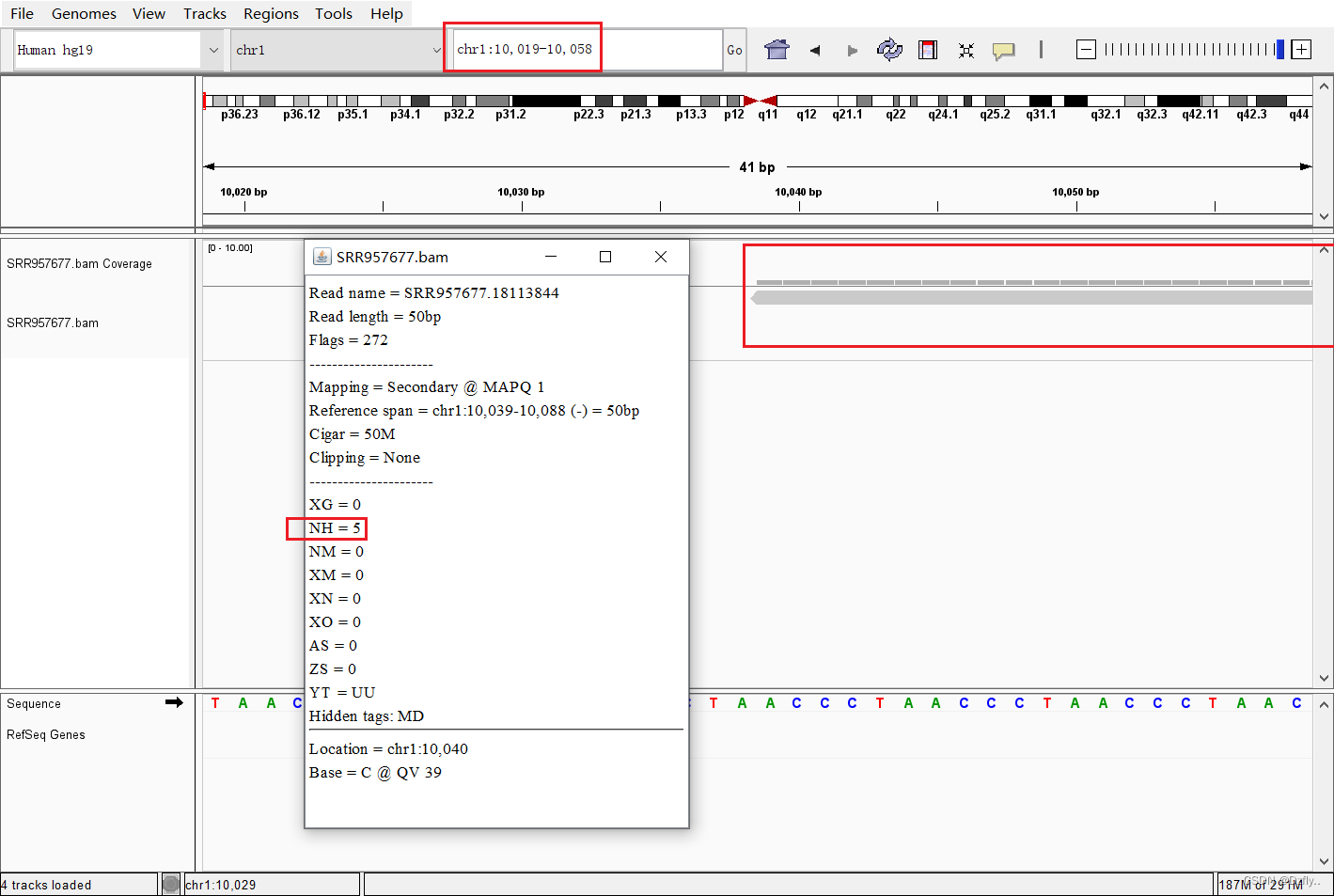
得到一个sam文件,结果如下:
@HD VN:1.0 SO:unsorted@SQ SN:22:20000001-21000000 LN:1000000@PG ID:hisat2 PN:hisat2 VN:2.0.0-beta1 0 22:20000001-21000000 397984 255 100M * 0 0 GCCTGTGAGGGAGCCCCGGACCCGGTCAGAGCAGGAGCCTGGCCTGGGGCCAAGTTCACCTTATGGACTCTCTTCCCTGCCCTTCCAGGAGCAGCTCACT IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:100 YT:Z:UU NH:i:12 16 22:20000001-21000000 398131 255 100M * 0 0 ATGACACACTGTACACACCAGGGGCCCTGTGCTCCCCAGGAAGAGGGCCCTCACTTGAAGCGGGGCCCGATGGCCGCCACGTGCCGGTTCATGCTCCCCT IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:80A19 YT:Z:UU NH:i:1 Zs:Z:80|S|rs5761598953 16 22:20000001-21000000 398222 255 100M * 0 0 TGCTCCCCTTGGCCCCGCCGATGTTCAGGGACATGGAGCGCTGCAGCAGGCTGGAGAAGATCTCCACTTGGTCAGAGCTGCAGTACTTGGCGATCTCAAA IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:16A83 YT:Z:UU NH:i:1 Zs:Z:16|S|rs26293644 16 22:20000001-21000000 398247 255 90M200N10M * 0 0 CAGGGACATGGAGCGCTGCAGCAGGCTGGAGAAGATCTCCACTTGGTCAGAGCTGCAGTACTTGGCGATCTCAAACCGCTGCACCAGGAAGTCGATCCAG IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:100 YT:Z:UU XS:A:- NH:i:15 16 22:20000001-21000000 398194 255 100M * 0 0 GGCCCGATGGCCGCCACGTGCCGGTTCATGCTCCCCTTGGCCCCGCCGATGTTCAGGGACATGGAGCGCTGCAGCAGGCTGGAGAAGATCTCCACTTGGT IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:17A26A55 YT:Z:UU NH:i:1 Zs:Z:17|S|rs576159895,26|S|rs26293646 0 22:20000001-21000000 398069 255 100M * 0 0 CAGGAGCAGCTCACTGAAATGTGTTCCCCGTCTACAGAAGTACCGTGATACACAGACGCCCCATGACACACTGTACACACCAGGGGCCCTGTGCTCCCCA IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:100 YT:Z:UU NH:i:17 0 22:20000001-21000000 397896 255 100M * 0 0 GTGGAGTAGATCTTCTCGCGAAGCACATTGCAGATGGTTGCATTTGGAACCACATCGGCATGCAGGAGGGACAGCCCCAGGGTCAGCAGCCTGTGAGGGA IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:31G68 YT:Z:UU NH:i:1 Zs:Z:31|S|rs5626622618 0 22:20000001-21000000 398150 255 100M * 0 0 AGGGGCCCTGTGCTCCCCAGGAAGAGGGCCCTCACTTGAAGCGGGGCCCGATGGCCGCCACGTGCCGGTTCATGCTCCCCTTGGCCCCGCCGATGTTCAG IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:61A26A11 YT:Z:UU NH:i:1 Zs:Z:61|S|rs576159895,26|S|rs26293649 16 22:20000001-21000000 398329 255 8M200N92M * 0 0 ACCAGGAAGTCGATCCAGATGTAGTGGGGGGTCACTTCGGGGGGACAGGGTTTGGGTTGACTTGCTTCCGAGGCAGCCAGGGGGTCTGCTTCCTTTATCT IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:100 YT:Z:UU XS:A:- NH:i:110 16 22:20000001-21000000 398184 255 100M * 0 0 CTTGAAGCGGGGCCCGATGGCCGCCACGTGCCGGTTCATGCTCCCCTTGGCCCCGCCGATGTTCAGGGACATGGAGCGCTGCAGCAGGCTGGAGAAGATC IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII AS:i:0 XN:i:0 XM:i:0 XO:i:0 XG:i:0 NM:i:0 MD:Z:27A26A45 YT:Z:UU NH:i:1 Zs:Z:27|S|rs576159895,26|S|rs2629364
sam文件转化成bam,然后排序,生成索引
samtools view -S ${i}.sam -b > ${i}.bam
####bam文件排序成sort.bam##
samtools sort ${i}.bam -n -o ${i}_sorted.bam
###Sort.bam建立index文件##samtools index ${i}_sorted.bam
####sort.bam进行比对得到bai和stat##
进行排序之后结果如下:
@HD VN:1.0 SO:coordinate
@SQ SN:chr1 LN:248956422
@SQ SN:chr2 LN:242193529
@SQ SN:chr3 LN:198295559
@SQ SN:chr4 LN:190214555
@SQ SN:chr5 LN:181538259
@SQ SN:chr6 LN:170805979
@SQ SN:chr7 LN:159345973
@SQ SN:chr8 LN:145138636
@SQ SN:chr9 LN:138394717
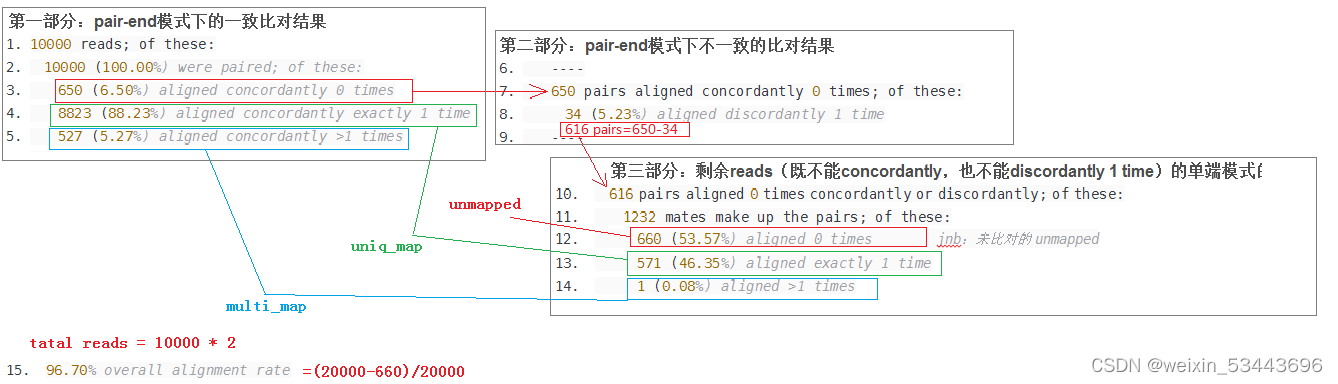
查看比对成功率
(base) [med-zhouh@login01 riboseq]$ cat *stat | grep %
76497603 + 0 mapped (91.97% : N/A)
91704826 + 0 mapped (93.17% : N/A)
72387422 + 0 mapped (91.84% : N/A)
88842249 + 0 mapped (91.83% : N/A)
去除重复再次比对
samtools markdup -r ${i}_sorted.bam ${i}.rmdup.bam
###再重复一个index##
samtools index ${i}.rmdup.bam
###继续比对一下##
samtools flagstat ${i}.rmdup.bam > ${i}.rmdup.stat
得到结果如下:
(riboseq) [med-zhouh@login01 riboseq]$ cat *.rmdup.stat | grep %
50411129 + 0 mapped (88.30% : N/A)
60915889 + 0 mapped (90.06% : N/A)
49874257 + 0 mapped (88.57% : N/A)
完成!




![转录组学习之序列比对(Hisat2)[学习笔记通俗易懂版]](https://img-blog.csdnimg.cn/e99c9df3c6ca436491401c75aafe334e.png)