参考文章:
RNAseq(4)–Hisat2进行序列比对及Samtools格式转化
RNA-seq(5):序列比对:Hisat2
hisat2比对软件将reads比对到参考基因组
hisat2比对
RNA-seq数据分析使用方法(陈建国 译)
转录组分析2——比对基因组
RNA-seq练习 第二部分
数据分析与解读(上)
转录组入门分析5_序列比对
1. 根据不同目的选择不同软件
RNA-Seq的主要目标是确定特定样品中RNA分子的序列、结构和丰度。
- 序列指的是A、C、G、U残基的特定顺序;序列可以用于识别一致的蛋白质编码基因、新基因或长的非编码RNA,一旦序列被确定,折叠成二级结构可以揭示分子的类型,如tRAN或miRNA。
- 结构是指基因结构【即启动子、内含子-外显子街头、5‘端和3’端非翻译区(UTR)级polyA位置】。二级结构提供互补的核苷酸配对和发夹或凸起的位置,三级结构提供分子的三维形状。
- 丰度指的是每个特定序列的数量值,作为绝对值和归一化的值。
RNA-seq的应用
- 蛋白质编码基因结构
- 新型蛋白质编码基因
- 基因表达的量化和比较
- 表达数量性状基因座
- 单细胞RNA-seq
- 融合基因
- 基因变异
- 长的非编码RNA
- 非编码小RNA
- 扩增产物测序
由以上RNA-seq的众多应用可以知道,RNA-seq可应用于多个方向的研究工作中。按照后边的参考索引,我应该主要使用参考基因组、参考转录组两种。将reads比对或作图到一个参考基因组或转录组允许我们估计reads源自哪里。
- 比对到参考基因组(genome)
1、将reads作图到基因组提供基因组的位置信息,这可以用于发现新的基因和转录本,以及用于量化表达。
2、面临困难:reads相对较短,数目以百万计,而基因组可能很大,并且包含非唯一的序列,如重复和假基因,降低这些区域的可作图性;比对程序必须应对由基因组变异和测序错误造成的不匹配和插入/缺失;许多生物在它们的基因中有内含子 ,因此RNA-seq读段非连续地比对到基因组。 - 比对到参考转录组(trans)
如果没有参考基因组可用,或者只想要量化已知的转录本,可将读段作图到一个转录组。
RNA-seq读段专化的剪接比对程序(spliced aligner)使用不同的方法来比对剪接的读段,包括执行一个初始化的比对,以发现外显子连接(exon junction),然后引导最终的比对。如果有基因组注释可用,比对程序可以使用它来放置剪接的读段。
当为RNA-seq研究选择一个比对程序时,主要考虑是否需要剪接比对(spliced alignment)。
- 如果生物体没有内含子,或对小分子RNA(microRNA)进行测序,完全可以使用连续地比对程序,如Bowtie或BWA,它们最初是为DNA开发的。
- 如果读段被作图到一个转录组而不是一个基因组,也可以使用这些比对程序。
- 如果RNA-seq读段被作图到含有内含子的基因组,则必须使用剪接比对程序,如TopHat、STAR或GSNAP。
由于二代测序的reads长度通常介于50~300个碱基,因此即便使用双端测序,也基本不可能覆盖完整的mRNA转录本,因此想直接用FastQ文件从头分析测到了哪些转录本需要非常复杂的分析和计算。好在通常情况下,公共数据库已经提供了测序样品的基因组和转录本的序列。因此我们只需要知道,每一条reads来自哪一条转录本就可以了,这个将reads与参考(Reference)基因组/转录组的序列进行比较和匹配的过程,我们通常称之为“比对”(文献中提到的read alignment和mapping通常说的都是这个)。
转录组测序的比对通常分为基因组比对和转录组比对两种,顾名思义,基因组比对就是把reads比对到完整的基因组序列上,而转录组比对则是把reads比对到所有已知的转录本序列上。如果不是很急或者只想知道已知转录本表达量,个人建议使用基因组比对的方法进行分析,理由如下:
① 转录组比对需要准确的已知转录本的序列,对于来自未知转录本(比如一些未被数据库收录的lncRNA)或序列不准确的reads无法正确比对;
② 与上一条类似,转录组比对不能对转录本的可变剪接进行分析,数据库中未收录的剪接位点会被直接丢弃;
③ 由于同一个基因存在不同的转录本,因此很多reads可以同时完美比对到多个转录本,reads的比对评分会偏低,可能被后续计算表达量的软件舍弃,影响后续分析(有部分软件解决了这个问题);
④ 由于与DNA测序使用的参考序列不同,因此不利于RNA和DNA数据的整合分析。
而上面的问题使用基因组比对都可以解决。
此外,值得注意的是,RNA测序并不能直接使用DNA测序常用的BWA、Bowtie等比对软件,这是由于真核生物内含子的存在,导致测到的reads并不与基因组序列完全一致(如下图所示),因此需要使用Tophat/HISAT/STAR等专门为RNA测序设计的软件进行比对。
HISAT2,取代Bowtie/TopHat程序,能够将RNA-Seq的读取与基因组进行快速比对。HISAT利用大量FM索引,以覆盖整个基因组。Index的目的主要使用与序列比对。由于物种的基因组序列比较长, 如果将测序序列与整个基因组进行比对,则会非常耗时。因此采用将测序序列和参考基因组的Index文件进行比对,会节省很多时间。以人类基因组为例,它需要48,000个索引,每个索引代表~64,000 bp的基因组区域。这些小的索引结合几种比对策略,实现了RNA-Seq读取的高效比对,特别是那些跨越多个外显子的读取。尽管它利用大量索引,但HISAT只需要4.3 GB的内存。这种应用程序支持任何规模的基因组,包括那些超过40亿个碱基的。
数据分析可以应用于不同目的的研究中,比如说找差异表达基因、或寻找新的可变剪切。如果找差异表达基因,只需要确定不同reads的数目,可以使用bowtie, bwa这类比对工具,或者是salmon这类align-free工具,并且后者的速度更快;如果需要找到新的isoform,或者RNA的可变剪切,看看外显子使用差异的话,就需要TopHat, HISAT2或者是STAR这类工具用于找到剪切位点。
基因组比对常用软件bwa,转录组比对软件常用bowtie2、hisat2等,其中有参考基因组的常用hisat2,没有参考基因组的常用bowtie2。
HISAT2 is a successor to both HISAT and TopHat2.
We recommend that the HISAT and TopHat2 users switch to HISAT2.
2. Hisat2安装
使用Miniconda3进行安装,步骤如RNA-seq流程学习笔记(3)。
3. Hisat2用法
(base) zexing@DNA:$ hisat2 -h
HISAT2 version 2.1.0 by Daehwan Kim (infphilo@gmail.com, www.ccb.jhu.edu/people/infphilo)
Usage:hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r> | --sra-acc <SRA accession number>} [-S <sam>]<ht2-idx> Index filename prefix (minus trailing .X.ht2).<m1> Files with #1 mates, paired with files in <m2>.Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).<m2> Files with #2 mates, paired with files in <m1>.Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).<r> Files with unpaired reads.Could be gzip'ed (extension: .gz) or bzip2'ed (extension: .bz2).<SRA accession number> Comma-separated list of SRA accession numbers, e.g. --sra-acc SRR353653,SRR353654.<sam> File for SAM output (default: stdout)<m1>, <m2>, <r> can be comma-separated lists (no whitespace) and can bespecified many times. E.g. '-U file1.fq,file2.fq -U file3.fq'.
主要参数:-x <hisat2-idx>参考基因组索引文件的前缀(目录及文件前缀)。-1 <m1>双端测序结果的第一个文件。若有多组数据,使用逗号将文件分隔。Reads的长度可以不一致。-2 <m2>双端测序结果的第二个文件。若有多组数据,使用逗号将文件分隔,并且文件顺序要和-1参数对应。Reads的长度可以不一致。-U <r>单端数据文件。若有多组数据,使用逗号将文件分隔。可以和-1、-2参数同时使用。Reads的长度可以不一致。–sra-acc <SRA accession number>输入SRA登录号,比如SRR353653,SRR353654。多组数据之间使用逗号分隔。HISAT将自动下载并识别数据类型,进行比对。-S <hit>指定输出的SAM文件(目录及名称)。-t/--time print wall-clock time taken by search phases-p/--threads <int> number of alignment threads to launch 指定进程数目(8/16)
4.关于Index的获得
1. [HISAT2官网下载](https://daehwankimlab.github.io/hisat2/)
HISAT2官网更新了新的网站,并且可以提供多个物种的Index直接下载进行序列比对,感觉更简单。


使用wget下载需要的索引:
#下载 H. sapiens, UCSC hg38 genome
wget https://cloud.biohpc.swmed.edu/index.php/s/hg38/download
#下载 H. sapiens, UCSC hg38 genome_tran
wget https://cloud.biohpc.swmed.edu/index.php/s/hg38_tran/download
#下载 M. musculus, UCSC mm10 genome
wget https://cloud.biohpc.swmed.edu/index.php/s/mm10/download
#关于genome 和genome-trans的选择,还需要继续学习。
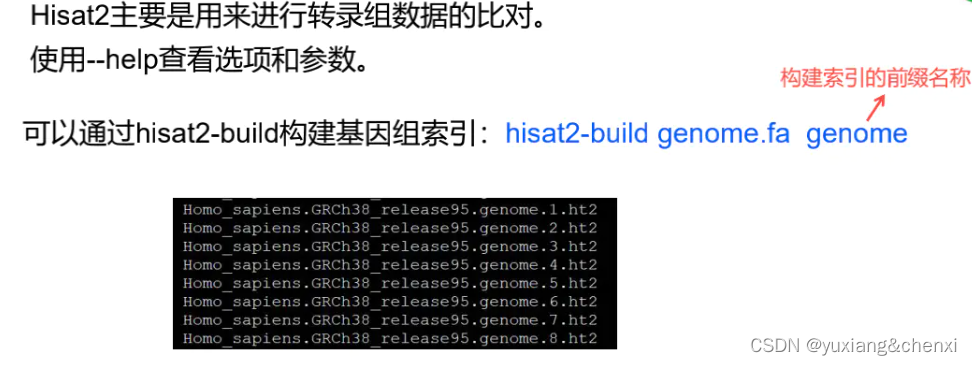
2. 使用Hisat2构建索引(暂未尝试)
有时候没有现成的index,我们就需要自己用HISAT2重新构建索引;包括外显子、剪切位点及SNP索引的建立。
# 其实hisat2-buld在运行的时候也会自己寻找exons和splice_sites,但是先做的目的是为了提高运行效率
extract_exons.py GCF_016699485.2_bGalGal1.mat.broiler.GRCg7b_genomic.gff > gg.exons.gtf
extract_splice_sites.py GCF_016699485.2_bGalGal1.mat.broiler.GRCg7b_genomic.gff > gg.splice_sites.gtf
# 建立index, 必须选项是基因组所在文件路径和输出的前缀
hisat2-build --ss hg19.splice_sites.gtf --exon hg19.exons.gtf genome/hg19/hg19.fa hg19
5. 使用Hisat2进行比对的记录
1. 对双端测序的m3110样品进行比对
#我的index使用同事构建的hisat2索引,索引所在目录:/f/xudonglab/zexing/reference/UCSC_mm10/hisat2_index/
#index中包含八个hisat2_index_mm10.*.ht2文件,因此index前缀写:hisat2_index_mm10
#我的fq.gz文件在目录:/f/xudonglab/zexing/projects/zhaoxiujuan/raw/
#选取双端测序的m3110进行测试
#提前新建比对后输出序列存放目录:/f/xudonglab/zexing/projects/zhaoxiujuan/aligned
#比对后的输出结果命名为:m3110.sam
#参数-p:设定线程数,选8
#参数-t:显示进程时间
(base) zexing@DNA:~/projects/zhaoxiujuan/raw$
hisat2 -t -p 8 -x /f/xudonglab/zexing/reference/UCSC_mm10/hisat2_index/hisat2_index_mm10 \
-1 /f/xudonglab/zexing/projects/zhaoxiujuan/raw/m3110_1.fq.gz \
-2 /f/xudonglab/zexing/projects/zhaoxiujuan/raw/m3110_2.fq.gz \
-S /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/m3110.sam
Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Time loading forward index: 00:00:02
Time loading reference: 00:00:00
#电脑显示到这里就一直没动静,这两天一直纠结是不是哪里出错卡住了,今天一直等它自己反应,才发现运行时间很长,最好是放到后台运行,长记性了!
Multiseed full-index search: 01:17:42
23824904 reads; of these:23824904 (100.00%) were paired; of these:1318623 (5.53%) aligned concordantly 0 times20713584 (86.94%) aligned concordantly exactly 1 time1792697 (7.52%) aligned concordantly >1 times----1318623 pairs aligned concordantly 0 times; of these:117263 (8.89%) aligned discordantly 1 time----1201360 pairs aligned 0 times concordantly or discordantly; of these:2402720 mates make up the pairs; of these:1550797 (64.54%) aligned 0 times752045 (31.30%) aligned exactly 1 time99878 (4.16%) aligned >1 times
96.75% overall alignment rate
Time searching: 01:17:43
Overall time: 01:17:46
(base) zexing@DNA:~/projects/zhaoxiujuan/raw$
#终于显示结束了。。。
最重要的是比对到基因组或是转录组上的比对率。人类基因组的比对率期望值是70-90%,会出现多个序列比对在有限的序列区称之为“多重比对序列”(multi-mapping reads);转录组上的比对率较低,由于未注释的转录本会被过滤且“多重比对序列”增加,由于同一个基因不同亚型共有外显子区。
确定代码:
hisat2 -t -p 8 -x /f/xudonglab/zexing/reference/UCSC_mm10/hisat2_index/hisat2_index_mm10 \
-1 /f/xudonglab/zexing/projects/zhaoxiujuan/raw/m3110_1.fq.gz \
-2 /f/xudonglab/zexing/projects/zhaoxiujuan/raw/m3110_2.fq.gz \
-S /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/m3110.sam
2. 使用shell script进行批量比对
关于shell脚本的使用,参考文章:Linux学习笔记-学习shell脚本的使用(持续更新)
(1)使用Vim编辑器编写Hisat2.sh脚本如下:
#! /bin/bash
# Program:
# This program is used for aligning of data of zhaoxiujuan
#History:
# 2020/06/02 zexing First releasefor i in Scr msh2 msh1 m3122 m3114 m3113 m3112 m3111 m3110 m3108
do
hisat2 -p 8 -x /f/xudonglab/zexing/reference/UCSC_mm10/hisat2_index/hisat2_index_mm10 \
-1 /f/xudonglab/zexing/projects/zhaoxiujuan/raw/"$i"_1.fq.gz \
-2 /f/xudonglab/zexing/projects/zhaoxiujuan/raw/"$i"_2.fq.gz \
-S /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/"$i".sam
done
(2)使用sh运行hisat2.sh脚本,并使用nohup command &将程序放入后台运行
- 关于后台执行命令,参考:linux后台执行命令:&和nohup
#先运行一下hisat2.sh脚本,看看是否能运行(之前没仔细看,放进后台发现名称错了!)
(base) zexing@DNA:~/projects$ sh hisat2.sh
Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
#报错跟单个运行一样,见上面参考文章;
#使用Ctrl+C中断程序;
^C(ERR): hisat2-align died with signal 2 (INT)
#使用nohup command &将shell script放入后台运行:
(base) zexing@DNA:~/projects$ nohup sh hisat2.sh &
[1] 8686
(base) zexing@DNA:~/projects$ nohup: ignoring input and appending output to 'nohup.out'
#使用cat命令查看后台程序运行情况:
(base) zexing@DNA:~/projects$ cat nohup.out
Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
(base) zexing@DNA:~/projects$ cd zhaoxiujuan/raw
(3)网友的shell script例子,留作学习:
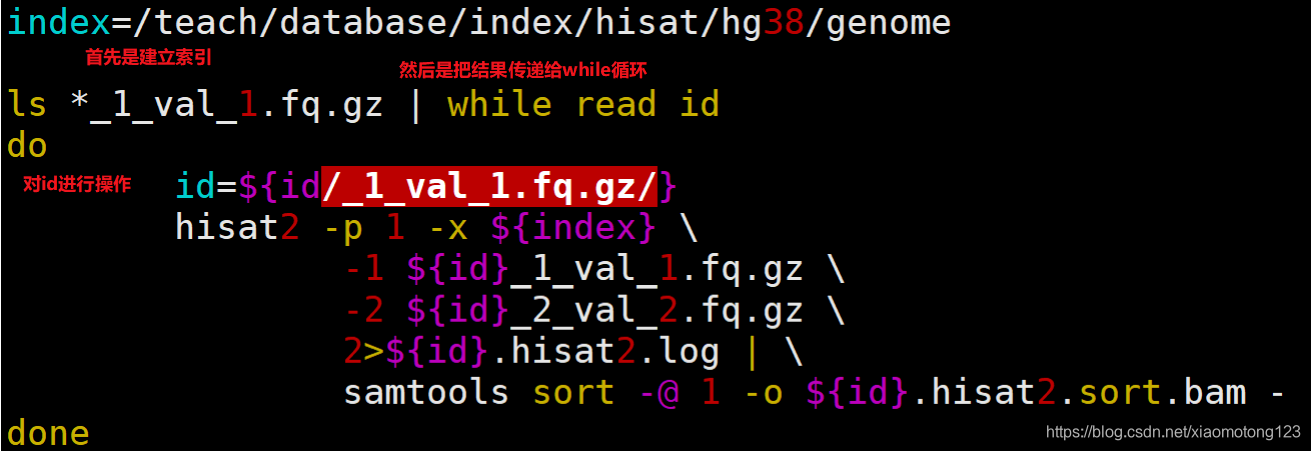 (4)查看nohup.out日志,检查结果运行情况:
(4)查看nohup.out日志,检查结果运行情况:
(base) zexing@DNA:~/projects/zhaoxiujuan/raw$ cat nohup.out
Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Time loading forward index: 00:00:03
Time loading reference: 00:00:00
Multiseed full-index search: 00:13:10
23385218 reads; of these:23385218 (100.00%) were paired; of these:1291253 (5.52%) aligned concordantly 0 times20890398 (89.33%) aligned concordantly exactly 1 time1203567 (5.15%) aligned concordantly >1 times----1291253 pairs aligned concordantly 0 times; of these:129434 (10.02%) aligned discordantly 1 time----1161819 pairs aligned 0 times concordantly or discordantly; of these:2323638 mates make up the pairs; of these:1518842 (65.36%) aligned 0 times722540 (31.10%) aligned exactly 1 time82256 (3.54%) aligned >1 times
96.75% overall alignment rate
Time searching: 00:13:11
Overall time: 00:13:14Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Time loading forward index: 00:00:03
Time loading reference: 00:00:00
Multiseed full-index search: 00:13:00
23065476 reads; of these:23065476 (100.00%) were paired; of these:1058976 (4.59%) aligned concordantly 0 times20824593 (90.28%) aligned concordantly exactly 1 time1181907 (5.12%) aligned concordantly >1 times----1058976 pairs aligned concordantly 0 times; of these:116284 (10.98%) aligned discordantly 1 time----942692 pairs aligned 0 times concordantly or discordantly; of these:1885384 mates make up the pairs; of these:1150090 (61.00%) aligned 0 times659877 (35.00%) aligned exactly 1 time75417 (4.00%) aligned >1 times
97.51% overall alignment rate
Time searching: 00:13:01
Overall time: 00:13:04Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Time loading forward index: 00:00:03
Time loading reference: 00:00:00
Multiseed full-index search: 00:12:35
22771010 reads; of these:22771010 (100.00%) were paired; of these:1056641 (4.64%) aligned concordantly 0 times20561945 (90.30%) aligned concordantly exactly 1 time1152424 (5.06%) aligned concordantly >1 times----1056641 pairs aligned concordantly 0 times; of these:122046 (11.55%) aligned discordantly 1 time----934595 pairs aligned 0 times concordantly or discordantly; of these:1869190 mates make up the pairs; of these:1141321 (61.06%) aligned 0 times651590 (34.86%) aligned exactly 1 time76279 (4.08%) aligned >1 times
97.49% overall alignment rate
Time searching: 00:12:35
Overall time: 00:12:39Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Time loading forward index: 00:00:03
Time loading reference: 00:00:00
Multiseed full-index search: 00:12:17
22736544 reads; of these:22736544 (100.00%) were paired; of these:1092643 (4.81%) aligned concordantly 0 times20492595 (90.13%) aligned concordantly exactly 1 time1151306 (5.06%) aligned concordantly >1 times----1092643 pairs aligned concordantly 0 times; of these:134834 (12.34%) aligned discordantly 1 time----957809 pairs aligned 0 times concordantly or discordantly; of these:1915618 mates make up the pairs; of these:1172633 (61.21%) aligned 0 times663598 (34.64%) aligned exactly 1 time79387 (4.14%) aligned >1 times
97.42% overall alignment rate
Time searching: 00:12:18
Overall time: 00:12:22Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Time loading forward index: 00:00:03
Time loading reference: 00:00:01
Multiseed full-index search: 00:13:12
23149628 reads; of these:23149628 (100.00%) were paired; of these:1173073 (5.07%) aligned concordantly 0 times20791982 (89.82%) aligned concordantly exactly 1 time1184573 (5.12%) aligned concordantly >1 times----1173073 pairs aligned concordantly 0 times; of these:158642 (13.52%) aligned discordantly 1 time----1014431 pairs aligned 0 times concordantly or discordantly; of these:2028862 mates make up the pairs; of these:1277159 (62.95%) aligned 0 times668842 (32.97%) aligned exactly 1 time82861 (4.08%) aligned >1 times
97.24% overall alignment rate
Time searching: 00:13:13
Overall time: 00:13:16Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Time loading forward index: 00:00:03
Time loading reference: 00:00:01
Multiseed full-index search: 00:13:55
23824904 reads; of these:23824904 (100.00%) were paired; of these:1308505 (5.49%) aligned concordantly 0 times21271723 (89.28%) aligned concordantly exactly 1 time1244676 (5.22%) aligned concordantly >1 times----1308505 pairs aligned concordantly 0 times; of these:130867 (10.00%) aligned discordantly 1 time----1177638 pairs aligned 0 times concordantly or discordantly; of these:2355276 mates make up the pairs; of these:1523157 (64.67%) aligned 0 times744694 (31.62%) aligned exactly 1 time87425 (3.71%) aligned >1 times
96.80% overall alignment rate
Time searching: 00:13:57
Overall time: 00:14:00Traceback (most recent call last):File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 210, in <module>reads_stat(args.read_file, args.read_count)File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 168, in reads_statfor id, seq in fstream:File "/f/xudonglab/zexing/miniconda3/bin/hisat2_read_statistics.py", line 44, in parser_FQif line[0] == '@':
IndexError: index out of range
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Warning: the current version of HISAT2 () is older than the version (2.1.0) used to build the index.Users are strongly recommended to update HISAT2 to the latest version.
Time loading forward index: 00:00:02
Time loading reference: 00:00:01
Multiseed full-index search: 00:12:57
23371257 reads; of these:23371257 (100.00%) were paired; of these:1170414 (5.01%) aligned concordantly 0 times21032864 (89.99%) aligned concordantly exactly 1 time1167979 (5.00%) aligned concordantly >1 times----1170414 pairs aligned concordantly 0 times; of these:151345 (12.93%) aligned discordantly 1 time----1019069 pairs aligned 0 times concordantly or discordantly; of these:2038138 mates make up the pairs; of these:1269312 (62.28%) aligned 0 times685662 (33.64%) aligned exactly 1 time83164 (4.08%) aligned >1 times
97.28% overall alignment rate
Time searching: 00:12:58
Overall time: 00:13:01

![转录组学习之序列比对(Hisat2)[学习笔记通俗易懂版]](https://img-blog.csdnimg.cn/e99c9df3c6ca436491401c75aafe334e.png)