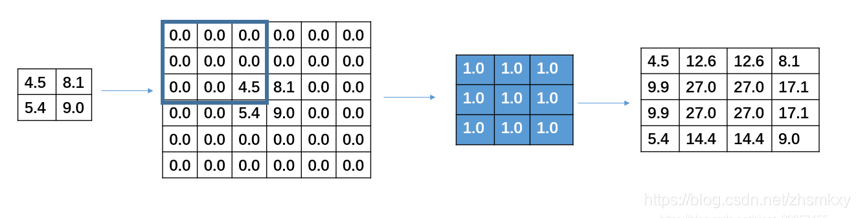
文章目录
- 反卷积的概念
- 反卷积的具体方式
- 反卷积预测RNA序列
- 知识背景
- 公式推导
- 亚硫酸氢盐测序
- 知识背景
- 公式推导
- R包的使用
- RNA测序数据分析
- 使用亚硫酸氢盐数据进行测序
反卷积的概念
由于许多组织样本不适合分解成单个细胞,因此不能利用单细胞RNA测序技术对它们的单个细胞进行测序。但是对于整个组织进行RNA测序是很容易的事情,而反卷积所做的就是由整体来探测局部,从而获知整个组织中的细胞类型,比如说了解实体肿瘤的特异性免疫细胞组成等等。
反卷积的具体方式
反卷积预测RNA序列
由于测序数据具有异方差性(对于不同的组织其线性模型中干扰项的方差不同)和呈离散分布的特性,因此线性模型不适合于进行预测。
负二项分布是统计学上一种描述在一系列独立同分布的伯努利试验中,失败次数到达指定次数(记为r)时成功次数的离散概率分布。
我们使用负二项模型来解释RNAseq数据中的二次均值 - 方差关系,并使用基因特异性方差估计。
知识背景
载体(Vector)
指在基因工程重组DNA技术中将DNA片段(目的基因)转移至受体细胞的一种能自我复制的DNA分子。三种最常用的载体是细菌质粒、噬菌体和动植物病毒。
基因文库
主要有两种基因文库:基因组文库和cDNA文库。
基因组文库:一个生物体的基因组DNA用限制性核酸内切酶部分酶切后,将酶切片段插入到载体DNA分子中,所有这些插入了基因组DNA片段的载体分子的集合体,将包含这个生物体的整个基因组,也就是构成了这个生物体的基因文库。
cDNA文库:是指某生物某发育时期所转录的全部 mRNA 经反转录形成的cDNA片段与某种载体连接而形成的克隆的集合体。
负二项分布
所有到失败r次时即终止的独立试验中,成功次数k的分布,在每次试验中,成功的概率为p,失败的概率为(1-p)。
- 均值:
μ = r 1 − p p \mu = r\frac{1-p}{p} μ=rp1−p
- 方差:
σ 2 = r 1 − p p 2 = μ p \sigma^2 = r\frac{1-p}{p^{2}} = \frac{\mu}{p} σ2=rp21−p=pμ
- α:
α = 1 − p p \alpha = \frac{1-p}{p} α=p1−p
-
离散参数
是指一个分布压缩和拉伸的程度,r越小,分布越集中,离散参数越大:
k = 1 r k = \frac{1}{r} k=r1 -
引入离散参数和α的方差:
α = μ ∗ k , σ 2 = α ( 1 + α ) k = μ + μ 2 ∗ k \alpha = \mu*k, \sigma^2 = \frac{\alpha(1+\alpha)}{k} = \mu + \mu^2*k α=μ∗k,σ2=kα(1+α)=μ+μ2∗k
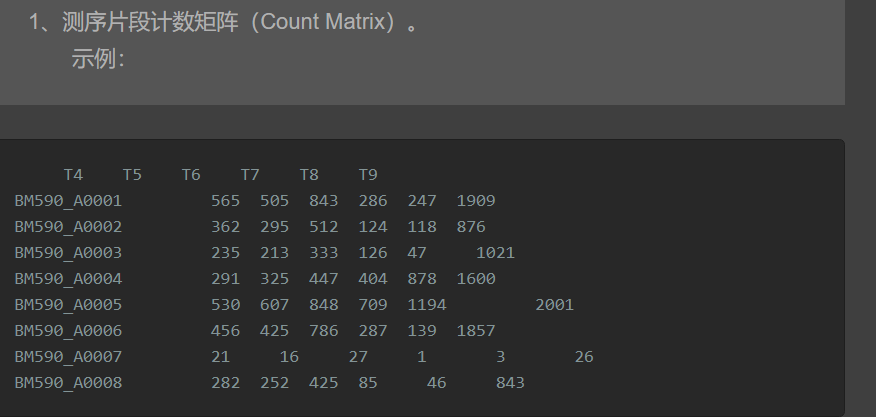
测序片段计数矩阵(Count Matrix)
第一列是样本名称,之后依次是对应不同基因组的技术,计数即每个样品中映射到每个感兴趣的基因组特征上的reads数目。
TMM
TMM,DESeq的前提假设都是大多数基因的表达是没有差异的,然后,基于这个假设根据均值,或者中值,比例等提出一个标准化的因子进行标准化。对于TMM这种方法,比例的计算是根据每次测序的数据和参考数据(libsize)进行比较。
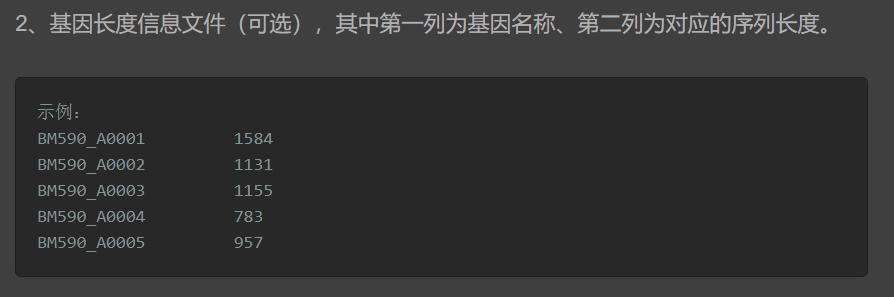
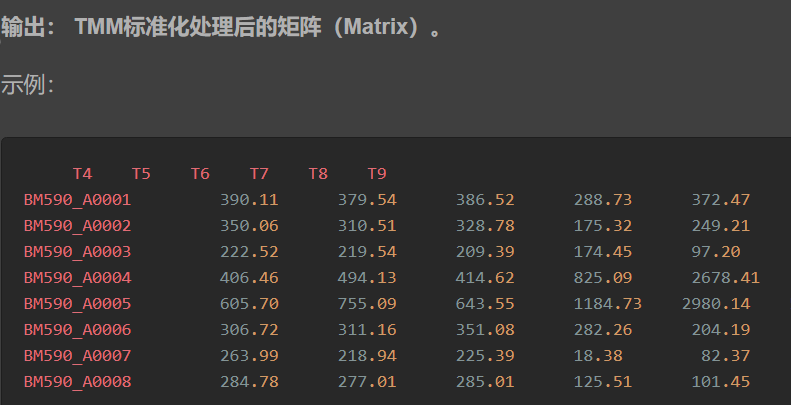
分析模块,采用edgeR的TMM(trimmed mean of M-values)方法对测序片段计数矩阵(Count Matrix)进行标准化处理。 如果不提供基因的长度信息文件,将只进行TMM标准化处理。果提供基因的长度信息文件,将使用TMM方法将Count数据转换为FPKM数据,输出FPKM矩阵。
输入:
null model
零模型,也就是所有变量之间不相关的模型,或者可以说是没有引入协变量的模型。
协变量
我想知道温度对于降水量的影响,但是海拔高度、经纬度、当地湿度等变量也会影响降水量。
那么,在我的研究中,温度F检验(ANOVA)就是自变量,降水量是应变量,而海拔高度、经纬度和当地湿度就是协变量。
F检验(ANOVA)
F分布是两个卡方分布(具有不同的自由度)的比值。
方差分析(ANOVA),又叫F检验,简单来说,就是求得F统计量(组间方差/组内方差),然后查F表,如果大于临界值(一般是0.05显著性水平下)则拒绝原假设,即组间具有显著性的差异。
F统计量 = 组间方差/组内方差
这里的方差等于平方和除以自由度,组间的自由度为(组数-1),组内自由度为组数*(样本量-1),此样本量可以不一样,所以方差分析各组的样本量可以不一样;
实际中我们的主要问题是看组间是否有差异,ANOVA告诉我们组间的差异不仅要看组间的波动,还要看组内的波动,如果组内波动太大的话,很可能不存在差异,只是组内的数据乱而已,当然组间的波动越大,则组间的差异越大。
Replicate
在生物科学中,复制品是正在分析的样品的精确副本,例如细胞,生物体或分子,在其上完成相同的程序。这通常是为了检查实验或程序错误。在没有错误的情况下,重复应该产生相同的结果。然而,重复不是假设的独立测试,因为它们仍然是相同的样本,因此不测试样本之间的变化。
技术重复:同一个样本,重复三次。此时样本量=1
生物重复:取多个样本,进行多次实验。此时样本量=3
细胞周期
细胞周期(cell cycle)是指细胞从一次分裂完成开始到下一次分裂结束所经历的全过程,分为间期与分裂期两个阶段。间期又分为三期、即DNA合成前期(G1期)、DNA合成期(S期)与DNA合成后期(G2期)。
- G1期(first gap) 从有丝分裂到DNA复制前的一段时期,又称合成前期,此期主要合成RNA和核糖体。该期特点是物质代谢活跃,迅速合成RNA和蛋白质,细胞体积显著增大。这一期的主要意义在于为下阶段S期的DNA复制作好物质和能量的准备。
- S期(synthesis) 即DNA合成期,在此期,除了合成DNA外,同时还要合成组蛋白。DNA复制所需要的酶都在这一时期合成。
- G2期(second gap)DNA合成后期,是有丝分裂的准备期。在这一时期,DNA合成终止,大量合成RNA及蛋白质,包括微管蛋白和促成熟因子等。
基因标识符
Entrze ID是美国NCBI数据库中的基因标识符,通常是由纯数字表示,比如人类TP53基因的Entrze ID是7157(注意,不同物种的基因ID是不同的);ensembl ID是欧洲生物信息数据库的基因标识符,都是以ENSG(ensembl gene)四个大写字母开始,后面跟着11位数字,所以ensembl ID的长度通常都是15位,比如人类TP53基因的ensembl ID是ENSG00000141510,值得注意的是ensembl ID不仅包含了两万多个蛋白质编码基因,同样也有很多的假基因、miRNA等,因此它的数量较多,有六万多个,比人类已知的基因数多得多;HGNC ID 是指由人类基因命名委员会(HUGO Gene Nomenclature Committee)指定的基因标识符,该委员会通常对基因赋予一个名字以及一个ID,比如人类TP53基因,其标准的symbol是TP53(相当于简称),标准的名称是tumor protein p53,HGNC ID 是11998。Refseq是美国NCBI提供的基因标准序列(参考序列)数据库,在该数据库中,人类TP53基因的ID是NG_017013。
公式推导
令k表示基因,其取值由1变化到m;h代表细胞样本数,其取值由1变化到n0;j代表每个样本内的细胞类型,其取值从1变化到d0;令Nh代表基因文库的大小;λhk代表样本h的reads里面能够映射到基因k的比例的期望值;w0hj代表样本h中的细胞类型j,需要注意的是,由于每个细胞样本中只包含一种细胞类型,因此对于某一个h,假设包含j*类型的细胞,则w0hj*=1,而对取其它值的j都为0;令log函数为负二项分布的链接函数,用g表示;令B0是个计数矩阵,行为细胞类型,列为基因的读数,那么B0jk表示细胞类型j包含的基因k的片段长度。
令:
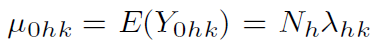
个人感觉上述公式计算的是样本h中基因k的长度的期望值。
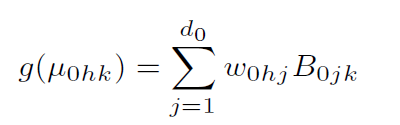
对于负二项分布,令φ0k为离散参数,而μ0hk可以看做是期望值,其方差满足:
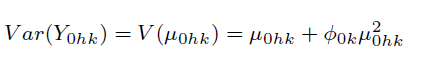
如果样本并不是全部样本,而是从1取到n1,令z1ij是样本i的协方差,有:
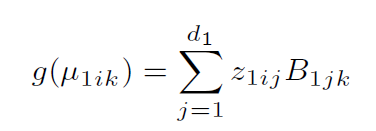
类似的,方差计算公式是:
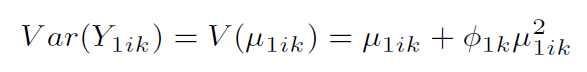
其中,φ0k可以通过软件算出来。
定义样本h的samplewise offset为αh,计算公式为log(Mh),其中Mh为样本h经过TMM处理之后的library size。设D0是null model的方差,而Df是full model的方差,F是F统计量。
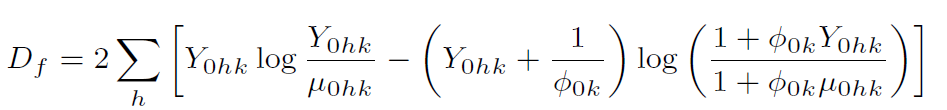
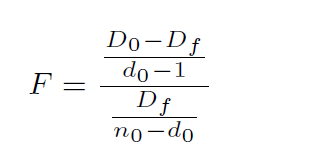
根据F统计量的大小,选取原始B0中的top r组基因,组成一个更小的新矩阵,称之为B0~,那么对于样本i,假设xji是其包含细胞类型j的比例,有:
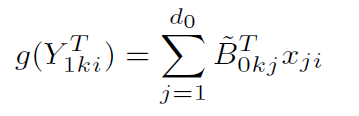
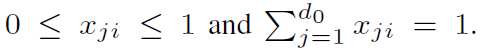
接下来计算Xi的对数极大似然,f是负二项分布的函数:
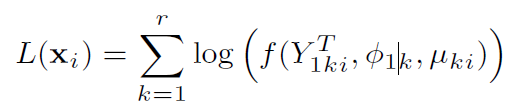
亚硫酸氢盐测序
由于数据里的标准定量采用的是甲基化的碱基比例的形式,因此要使用Logit函数做预处理。同时,统计检验量更改为对数似然比,而不是F统计量。
选取一定的特征来构建投影矩阵
知识背景
亚硫酸盐定序
亚硫酸盐定序是一种利用亚硫酸盐处理,测定DNA甲基化情形的方法。DNA甲基化是最早被发现的表观遗传标记,也是被研究最为深入的表观遗传改变。原理在于亚硫酸盐可使DNA上的胞嘧啶(C)转变为尿嘧啶(U),同时已受甲基化的5-甲基胞嘧啶则不受影响。如此一来,就可以使实验者得知DNA序列上甲基化的情形。 对动物而言,DNA甲基化主要是指对CpG位点中胞嘧啶的五号碳上增加一个甲基的过程。DNA位点的甲基化可能对该基因的转录活性起到抑制作用。
对数似然比检验
似然比检验的思想是:如果参数约束是有效的,那么加上这样的约束不应该引起似然函数最大值的大幅度降低。也就是说似然比检验的实质是在比较有约束条件下的似然函数最大值与无约束条件下似然函数最大值。
甲基化
NA甲基化是最早发现的修饰途径之一,可能存在于所有高等生物中。DNA甲基化导致某些区域DNA构象变化,从而影响了蛋白质与DNA的相互作用,抑制了转录因子与启动区DNA的结合效率,能关闭某些基因的活性,去甲基化则诱导了基因的重新活化和表达。DNA甲基化的主要形式为5-甲基胞嘧啶,N6-甲基腺嘌呤和7-甲基鸟嘌呤。在真核生物中,5-甲基胞嘧啶主要出现在CpG和CpXpG中,原核生物中CCA/TGG和GATC也常被甲基化;没有甲基化的胞嘧啶发生脱氨基作用,就可能被氧化成为U,被DNA修复系统所识别和切除,恢复成C;已经甲基化的胞嘧啶发生脱氨基作用, 它就变为T, 无法被区分。因此, CpG序列极易丢失,甲基化胞嘧啶极易在进化中丢失,所以,高等真核生物中CG序列远远低于其理论值;哺乳类基因组中约存在4万个CG islands,大多位于转录单元的5’区。
CpG位点
CpG位点是指DNA的某个区域,其上的碱基序列以胞嘧啶接着鸟嘌呤出现。“CpG”是“—C—磷酸—G—”的缩写,指磷酸二酯键连接了胞嘧啶和鸟嘌呤,其中C位于5’端而G位于3’端。 在CpG位点中的胞嘧啶可以被甲基化为5-甲基胞嘧啶。
公式推导
令Y0hk和Y1ik代表甲基化比例,对数似然比检验公式为:
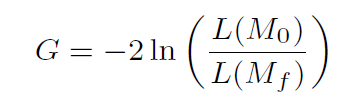
对数似然函数
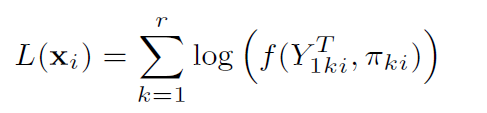
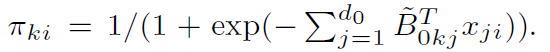
R包的使用
RNA测序数据分析
-
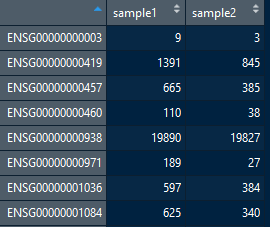
读取deconvSeq包中提供的两个单个样本的计数文件,并将它们组合成一个count matrix。
file1 = system.file("extdata","sample1_genecounts.txt", package="deconvSeq") file2 = system.file("extdata","sample2_genecounts.txt", package="deconvSeq") countmat = getrnamat(filnames=c(file1,file2),sample.id=c("sample1","sample2"))
-
从data_celltypes_rnaseq中加载单个细胞类型的数据,其中包括
cnts.celltypes,每种细胞类型的计数矩阵;针对cnts.celltypes的设计矩阵design.rnaseq;dge.celltypes即cnts.celltypes对应的dgelist对象;sample.id.rnaseq是列的名字。data("data_celltypes_rnaseq")-
cnts.celltypes
500个基因,64种细胞类型,具体的数值是细胞类型包含的基因的长度。
-
-
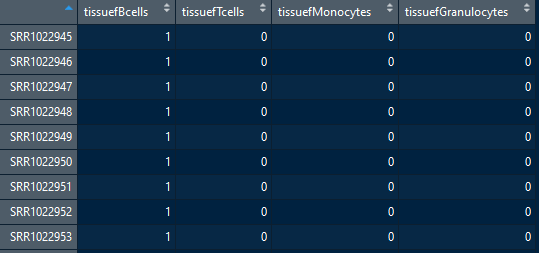
design.rnaseq
4个组织样本,每个组织样本对应包含64种细胞类型中的若干种,具体的数值是包含为1,不包含为0。并且每个细胞只会在每个样本中出现一次。
-
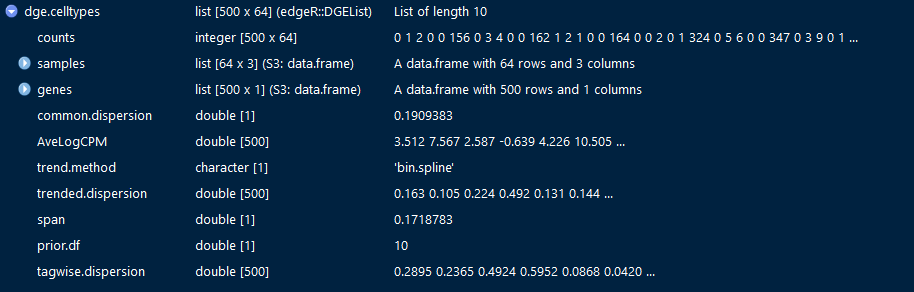
dge.celltypes
跟cnts.celltypes差不多,多了一些补充的数据,比如每种细胞类型的library size。
-
sample.id.rnaseq
就是64种细胞类型的名称
-
dge.celltypes的计算过程
dge.celltypes = getdge(cnts.celltypes, design.rnaseq, ncpm.min=1, nsamp.min=4) -
计算投影矩阵
b0,如果有一组预定的特征基因,可以在中指定sigg。默认sigg值为NULL。set.seed(1234) b0 = getb0.rnaseq(dge.celltypes, design.rnaseq, ncpm.min=1, nsamp.min=4, sigg=NULL)
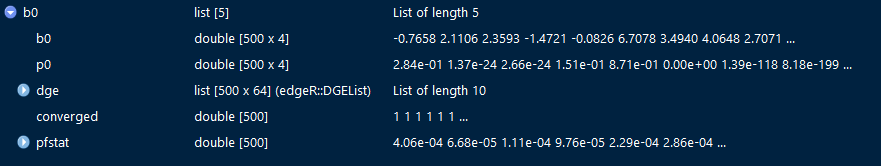
-
根据F统计量,选取top 50组基因构成签名集也就是预测细胞类型时所使用的特征基因。
resultx1 = getx1.rnaseq(NB0=50,b0,dge.celltypes)
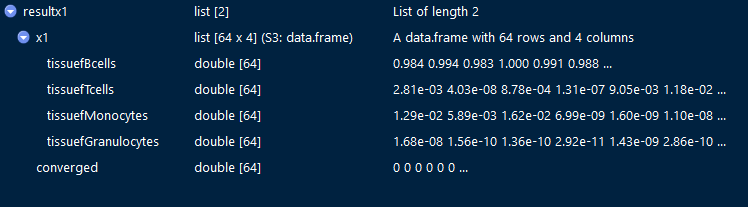
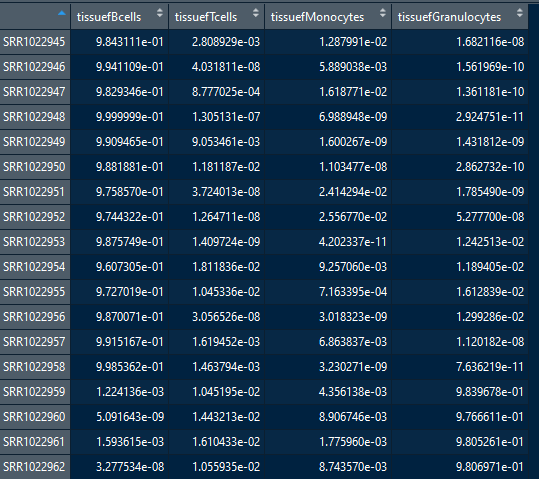
注意到每一行的和都是1,因此第x行y列的数值对应的生物学含义应该是第y个样本包含第x种细胞类型的概率。
-
计算相关系数
x2 = as.data.frame(design.rnaseq,row.names=sample.id.rnaseq)
cr = getcorr(resultx1$x1,x2)
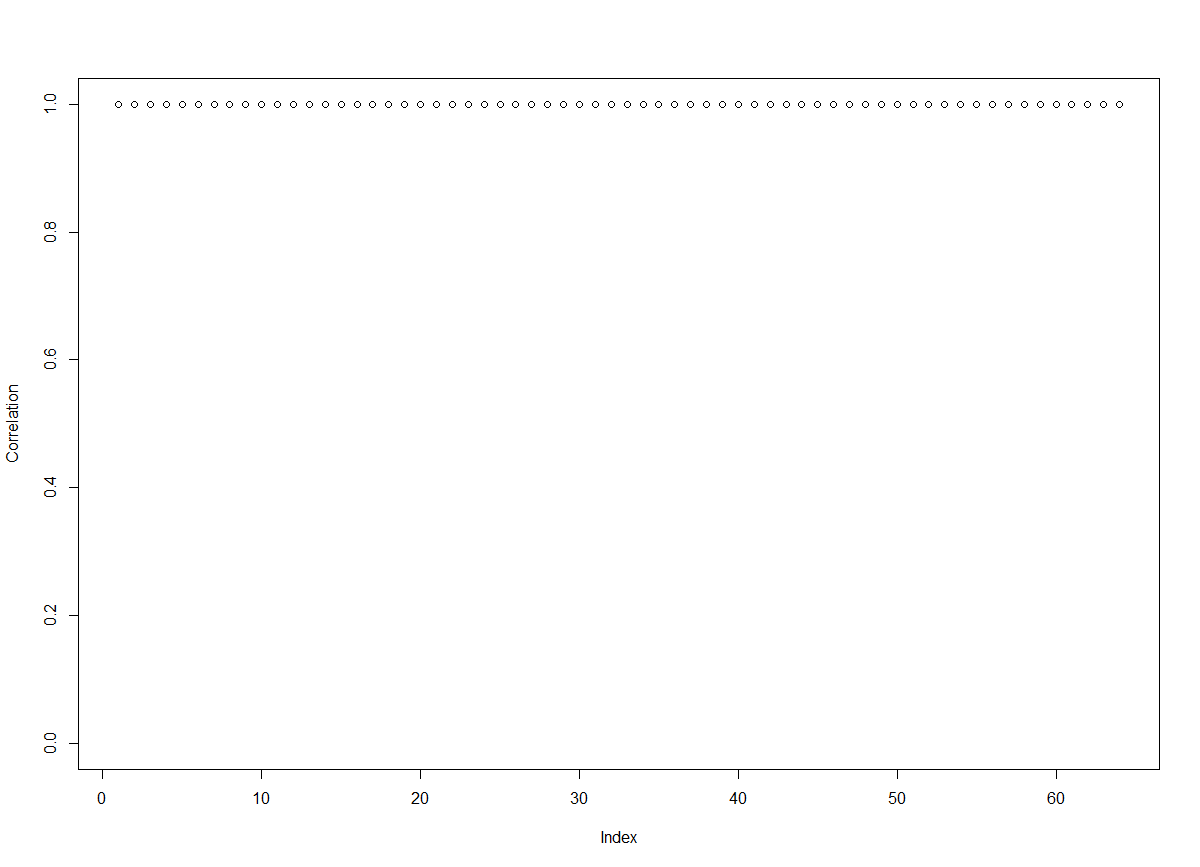
plot(cr, ylim=c(0,1), ylab=“Correlation”)
相关系数是按行计算的,因此最终结果是长为64的向量。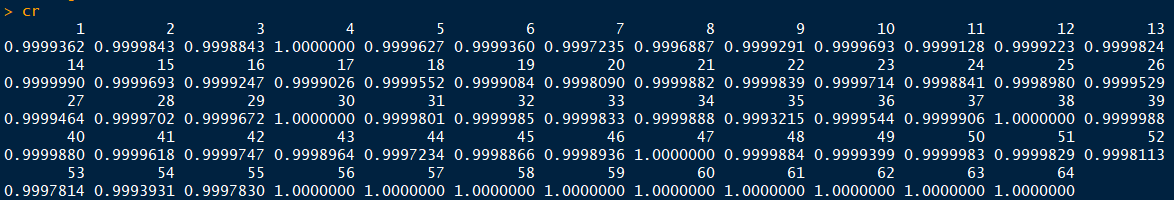
### 实际单细胞RNA测序分析- 加载数据```R
data("data_scrnaseq")
-
cnts.scrnaseq是一个计数矩阵,列指的是基因类型,行指的是两种细胞类型的若干样本。prep_scrnaseq用于对rnaseq进行质量控制,genenametype参数用于指定基因名字的类型,cellcycle表示细胞周期,count.threshold表示要过滤掉的数据的平均读数的阈值cnts.sc = prep_scrnaseq(cnts.scrnaseq, genenametype = "hgnc_symbol",cellcycle=NULL,count.threshold=0.05)
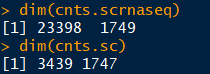
基因和细胞都有被过滤的,细胞基因表达数过少或基因丰度过低都是剔除条件。
-
过滤细胞周期阶段“G1”
cnts.sc.G1 = getcellcycle(cnts.sc,"G1")
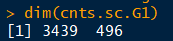
可以看到这次只对细胞进行了过滤。
-
等量从HuTreg和HuTconv这两种细胞类型中选择数据,将数据划分为训练集和验证集。design.sc意在揭示组织样本和细胞类型之间的关系。
cnts.sc.G1.train = cnts.sc.G1[,c(which(substr(colnames(cnts.sc.G1),3,6)=="Tcon")[1:250],which(substr(colnames(cnts.sc.G1),3,6)=="Treg")[1:150])] cnts.sc.G1.valid = cnts.sc.G1[,-which(colnames(cnts.sc.G1) %in% colnames(cnts.sc.G1.train))] tissue.sc = substr(colnames(cnts.sc.G1.train),3,6) names(tissue.sc) = colnames(cnts.sc.G1.train) sample.id.sc = colnames(cnts.sc.G1.train) design.sc = model.matrix(~-1+as.factor(tissue.sc)) colnames(design.sc) = levels(as.factor(tissue.sc)) rownames(design.sc) = names(tissue.sc) design.sc = design.sc[colnames(cnts.sc.G1.train),]
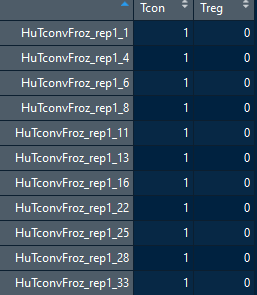
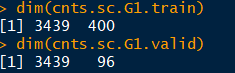
- 从计数矩阵
cnts.sc.G1.train,获取DGEList对象dge.sc,和投影矩阵b0.sc。
dge.sc = getdge(cnts.sc.G1.train,design.sc,ncpm.min=1, nsamp.min=4, method="bin.loess")
b0.sc = getb0.rnaseq(dge.sc, design.sc, ncpm.min=1, nsamp.min=4)
- 将投影矩阵应用于验证集
cnts.sc.G1.valid。
tissue_s.sc = substr(colnames(cnts.sc.G1.valid),3,6)
names(tissue_s.sc) = colnames(cnts.sc.G1.valid)
sample.id_s.sc = colnames(cnts.sc.G1.valid)
design_s.sc = model.matrix(~-1+as.factor(tissue_s.sc))
colnames(design_s.sc) = levels(as.factor(tissue_s.sc))
rownames(design_s.sc) = names(tissue_s.sc)
design_s.sc = design_s.sc[colnames(cnts.sc.G1.valid),]
dge_s.sc = getdge(cnts.sc.G1.valid, design_s.sc, ncpm.min=1, nsamp.min=4, method="bin.loess")
resultx1_s.sc = getx1.rnaseq(NB0=1500,b0.sc, dge_s.sc)
- 检查预测结果与实际细胞类型的相关性。
x2 = as.data.frame(design_s.sc,row.names=sample.id_s.sc)
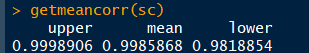
sc = getcorr(resultx1_s.sc$x1,x2)
getmeancorr(sc)
使用亚硫酸氢盐数据进行测序
-
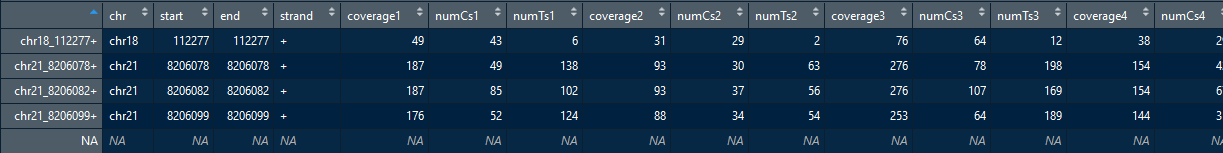
加载样本数据,获取甲基化矩阵
file1 = system.file("extdata","sample1_methratio.txt", package="deconvSeq") file2 = system.file("extdata","sample2_methratio.txt", package="deconvSeq") methmat = getmethmat(filnames=c(file1,file2), sample.id=c("sample1","sample2"))
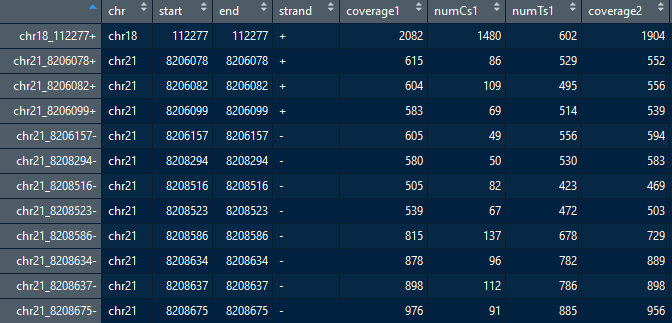
第一列表示cpg测序位点
chr列表示DNA位于哪个染色体上
strand为”-“,表示CpG在负链上;为”+”,表示CpG在正链上
numCs为C出现的次数,后面的数字应该表示的是细胞类型,共24种
numTs为T出现的次数
coverage列指的是,覆盖度 ,可以理解成是C和T的总数,而且coverage确实是这两者之和。因为亚硫酸氢钠处理后, 正常的C会转成T,甲基化的C不改变,所以用numCs除以coverage,就可以得到该位点发生甲基化的概率
-
加载
data_celltypes_rrbs单个细胞类型(T细胞,B细胞,单核细胞,粒细胞)的实例数据。这包括celltypes.rrbs,数据中的细胞类型;design.rrbs,设计矩阵;methmat,甲基化矩阵;sample.id.rrbs,样本ID。data("data_celltypes_rrbs")- celltypes.rrbs

- design.rrbs
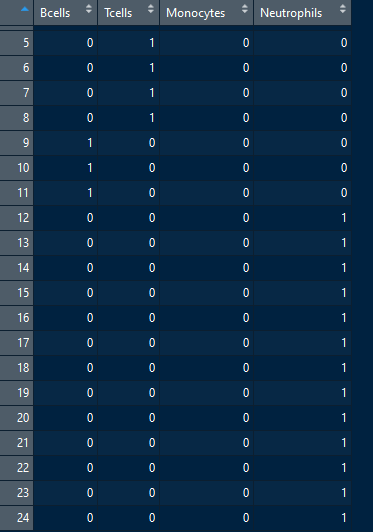
24个样本,每个样本包含一种细胞
- sample.id.rrbs,24个样本的id

-
计算投影矩阵
b0。可以指定预定的签名CpG集sigg。默认为siggNULL。set.seed(1234) b0 = getb0.biseq(methmat, design.rrbs, sigg=NULL)

- β值=来自甲基化珠粒类型的强度值/(来自甲基化的强度值+来自未甲基化珠粒类型的强度值+ 100)
- p值大于0.01的β值被认为低于最小强度,阈值显示为“NA”

-
mean.meth.diff:甲基化平均差异程度
-
根据F统计量,选取top 250组cpg构成签名集也就是预测细胞类型时所使用的特征cpg。
resultx1 = getx1.biseq(NB0=250,b0,methmat,sample.id.rrbs,celltypes.rrbs)
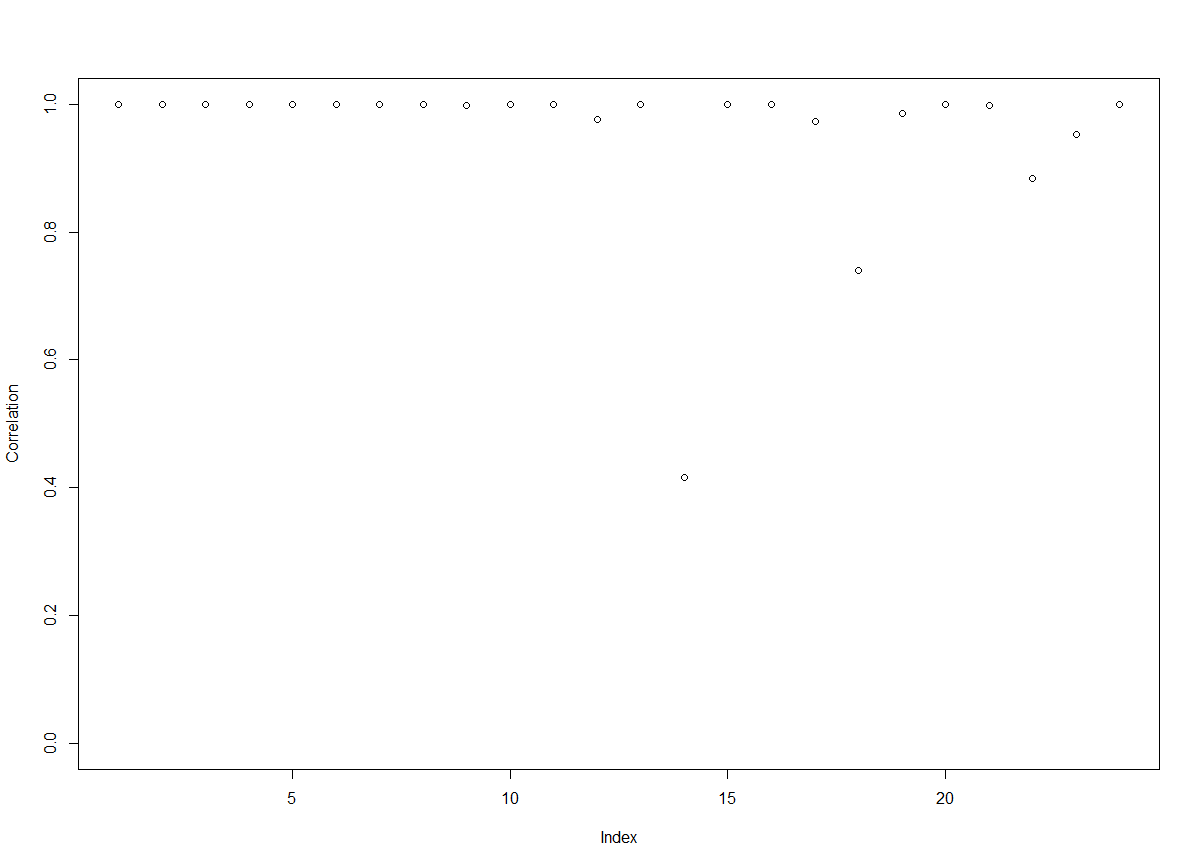
-
将预测结果与实际细胞类型
x2进行比较x2 = as.data.frame(design.rrbs,row.names=sample.id.rrbs) cr=getcorr(resultx1$x1,x2) plot(cr, ylim=c(0,1), ylab="Correlation")
-
加载整个组织样本的数据
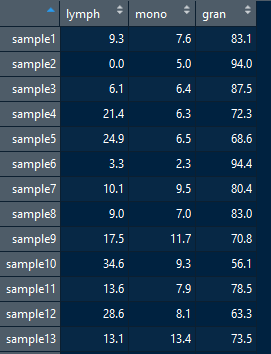
data_tissue_rrbs。这包括methmat.tissue,全血的甲基化矩阵;sample.id.tissue,样品ID;cbc.rrbs,实际细胞组成的矩阵。data("data_tissue_rrbs")- methmat.tissue
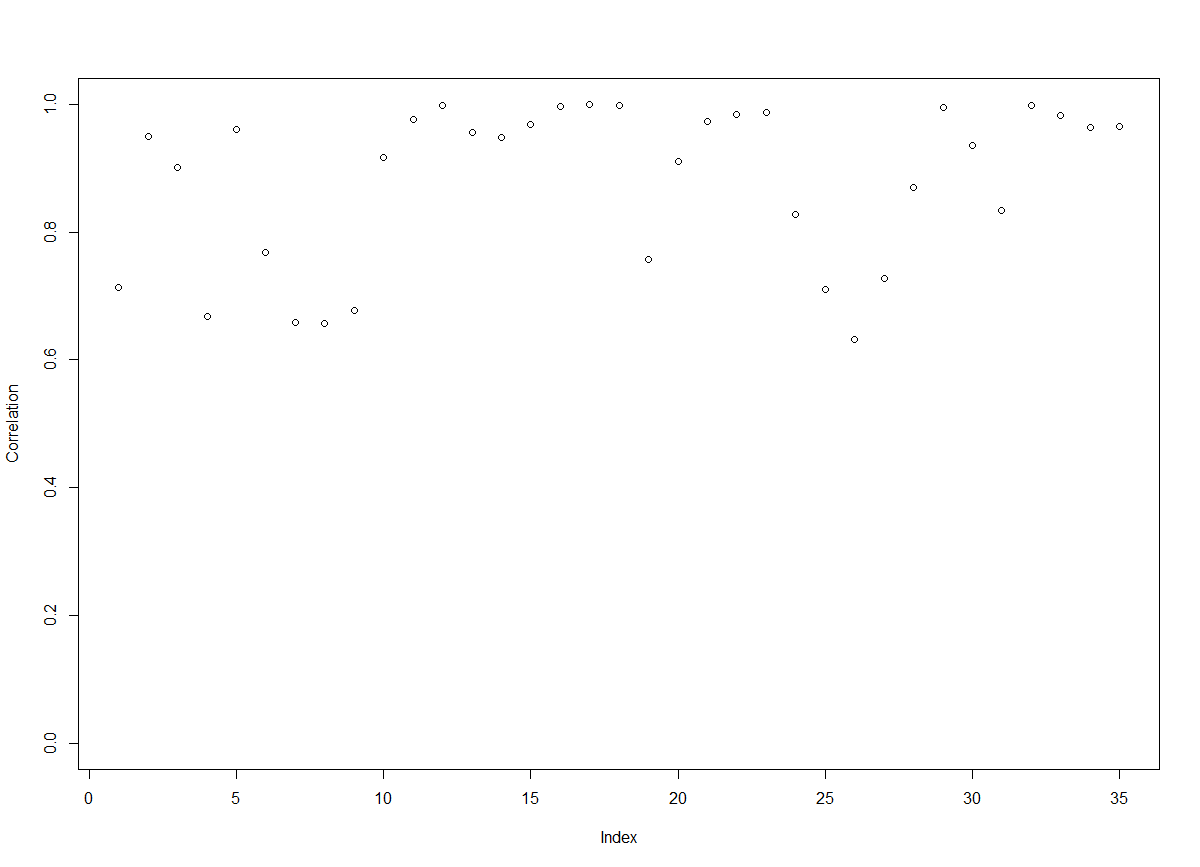
共35个样本
- sample.id.tissue

- cbc.rrbs,细胞类型分别为淋巴细胞,单核细胞,中性粒细胞
-
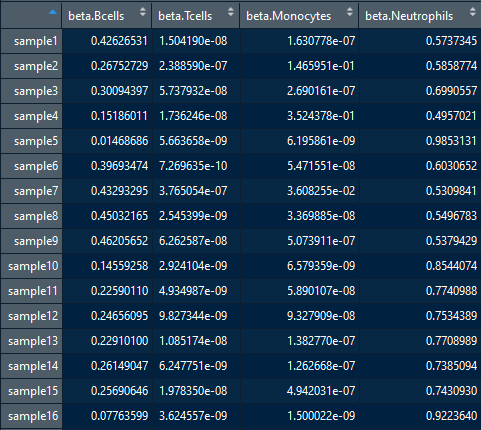
使用来自投影矩阵的250个特征CpG
b0,我们计算整个组织样本的预测细胞类型resultx1.tissue。resultx1.tissue = getx1.biseq(NB0=250,b0,methmat.tissue,sample.id.tissue,celltypes.rrbs)
-
把预测的细胞类型混合物与组织样品的已知细胞类型混合物进行比较。(注意到淋巴细胞等于T细胞+B细胞)
x1 = cbind(lymph=resultx1.tissue$x1[,1]+resultx1.tissue$x1[,2], mono = resultx1.tissue$x1[,3], gran = resultx1.tissue$x1[,4]) x2 = as.matrix(cbc.rrbs/100) cr = getcorr(x1,x2) plot(cr, ylim=c(0,1), ylab="Correlation")