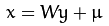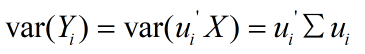在进行统计学分析中往往面临着比较难以抉择的权衡。以农学研究为例,在实验设计时,考虑到研究结论更能反应作物真实状态下的农艺性状,研究人员会尽可能的纳入较多的指标,但是,随着而来的是铺天盖地的数据让人难以下手,主成分分析(principal component analysis,PCA)便很好的解决了这一问题。
在生物学相关(因为我主要从事生物学研究 ^ _^)领域,PCA应用范围极广。光我接触过的便有数种:群体遗传学遗传成分的划分、代谢组学关键化合物的分离、群落学不同群落差异的评估、环境DNA组分的划分……
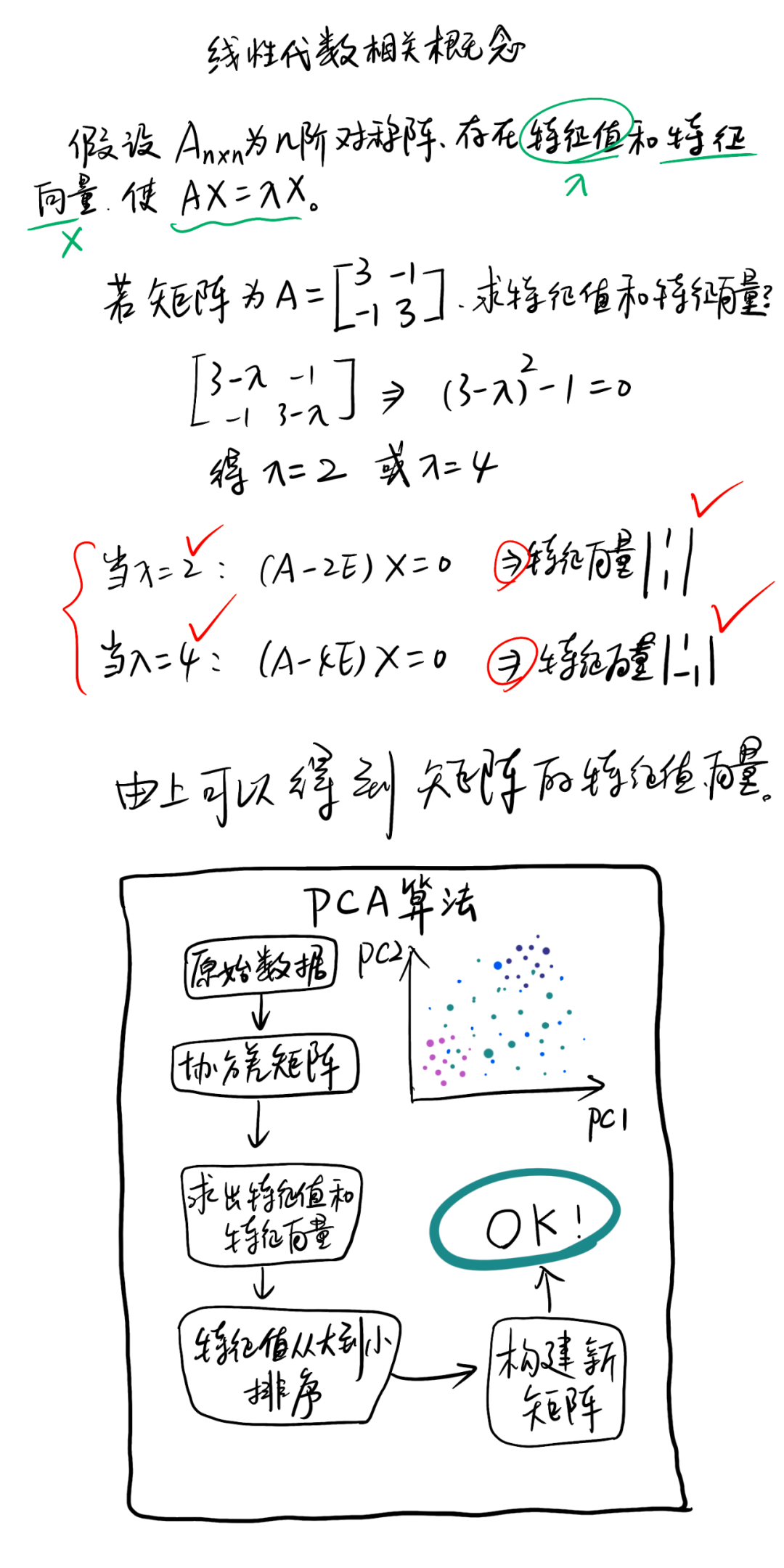
1 PCA原理
假设我们分别调查了几个群落的物种丰富度和各物种的多度,正如上述所言,就算只对三个群落进行调查,每个群落三个生物学重复,但是哪怕是群落构成单一如农田生态系统也有近百种物种构成,这时我们拿到的数据就是一个 9 × 100 9\times100 9×100的数据矩阵。
为了方便理解,假设我调查了 i i i个群落,其中物种最多的群落有 j j j个物种,这时我们就得到了下面的一个数据矩阵 A i × j A_{i\times j} Ai×j:
A i × j = [ x 11 x 12 … … x 1 j x 21 x 22 … … x 2 j . . … … . x i 1 x i 2 … … x i j ] A_{i\times j}= \begin{bmatrix} x_{11} & x_{12}& …… & x_{1j}\\ x_{21} & x_{22}& …… & x_{2j}\\ . & . & …… & . \\ x_{i1} & x_{i2} & …… & x_{ij} \\ \end{bmatrix} Ai×j= x11x21.xi1x12x22.xi2……………………x1jx2j.xij
而每个物种即构成了一个这个矩阵的列向量 x → j \overrightarrow x_j xj:
( x → 1 , x → 2 , … … , x → j ) (\overrightarrow x_1,\overrightarrow x_2,……,\overrightarrow x_j) (x1,x2,……,xj)
避开晦涩的数学推导过程,通俗的以几何图形的方式理解。在这个物种构成矩阵,可形成一个 j j j维的空间,每个物种的列向量 x → j \overrightarrow x_j xj为其所在维度的坐标系:
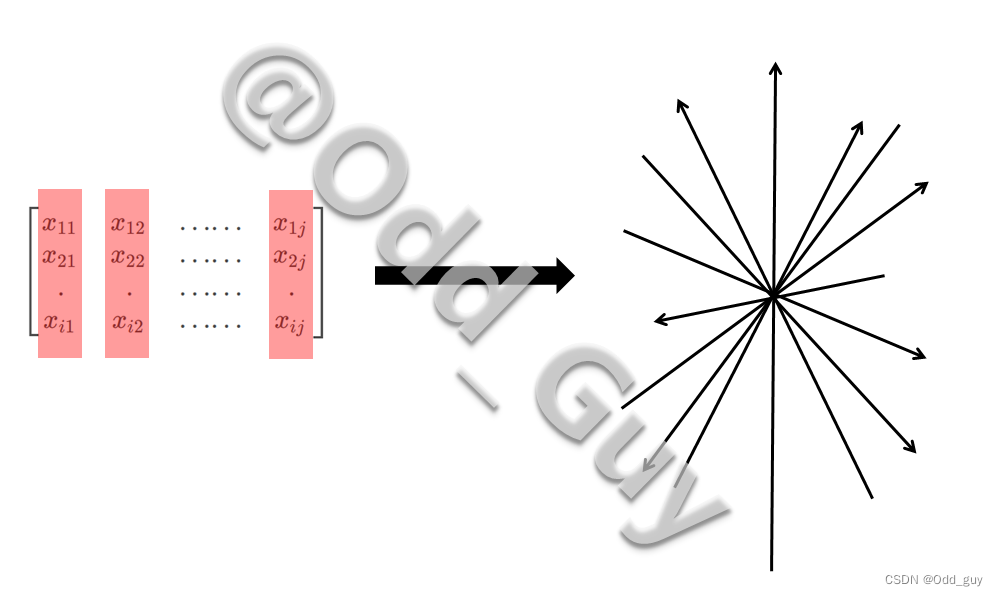
每个样本构成的行向量:
( x → 1 , x → 2 , … … , x → i ) (\overrightarrow x_1,\overrightarrow x_2,……,\overrightarrow x_i) (x1,x2,……,xi)
可看作是在此 j j j维空间内的 i i i个点,其对应的物种构成即为 j j j维坐标:

通过对每个向量构成元素的离散度(方差 S 2 S^2 S2)进行比较,离散度最大的向量对组间变异(即群落构成差异)的解释率最高,那么这个向量 x → n \overrightarrow x_n xn即为主成分1(PC1):
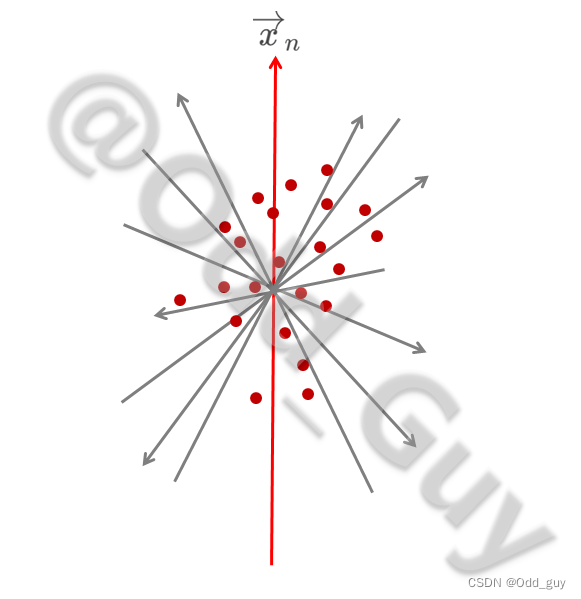
为避免各解释向量间的交互效应,过PC1轴取其正交平面 α \alpha α。在 α \alpha α平面内再次取离散度最大的向量轴 x → m \overrightarrow x_m xm作为主成分2(PC2):
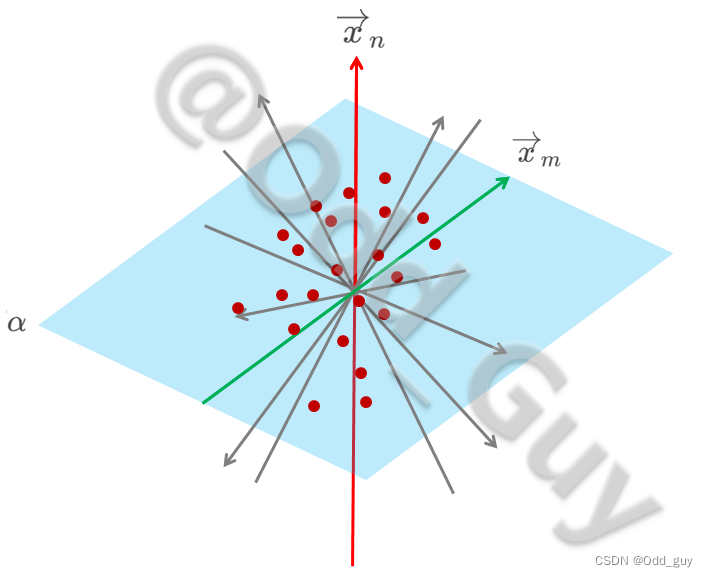
最后将结果在二维的平面展示就是我们经常看到的PCA可视化结果:
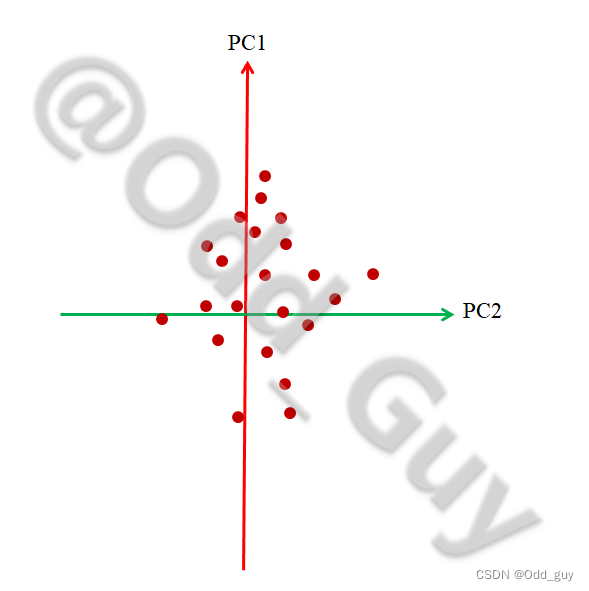
通过此方法即可快速找出数据矩阵中对组间变异解释度(贡献率)最高的几个指标,在此例子中就是对各群落结构差异贡献最大的几个物种。同理,将此思路推广到其他几个应用领域即为:对农产品品质差异影响最大的农艺性状;对代谢组差异贡献最大的代谢物……
2 实战
vegan包是生态学分析中较为常用的包,特别是各种排序图绘制功能(包括PCA),但缺点是绘制的图可能不及ggplot系列(没错,ggplot也有针对PCA的功能)美观。
2.1 vegan包
此时使用的数据是一个模拟的宏基因组数据,三个样点,每个样点20个重复,总计60个样本。
首先加载包、读取数据:
#加载包,前两个为依赖包
library(permute)
library(lattice)
library(vegan)#1.
#设置工作环境,读取数据
getwd()
setwd("D:/dir/CSDN/PCA/")#读取数据
data<-read.csv("example.csv",header=T)head(data)
#site otu_1 otu_2 otu_3 otu_4 otu_5 otu_6 otu_7 otu_8 otu_9 otu_10 otu_11 otu_12 otu_13 otu_14 otu_15 otu_16 otu_17 otu_18 otu_19 otu_20 otu_21 otu_22#site_1 1 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 2 0 0 0 0 1#site_1 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 2 0 0 0 0#site_1 0 6 0 0 0 0 0 1 0 0 0 0 0 0 0 0 1 0 0 0 0 0#site_1 1 0 6 1 6 1 5 0 0 1 1 0 1 0 0 1 1 0 0 0 0 0#site_1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 4 0 0 0 0 0#site_1 2 5 0 55 7 0 0 0 0 0 0 0 2 0 0 0 6 0 0 2 0 4
根据数据维度对数据进行切片降维:
#2.
#数据分析#查看数据维度,因为降维过程不需要样点信息列
dim(data)
#输出为:
#[1] 60 94#根据前一步输出提取数据的主要部分,并降维
data_pca<-rda(data[,2:94])#查看主要结果
summary(data_pca)
由于是模拟数据的缘故,解释率可能会比较奇怪:
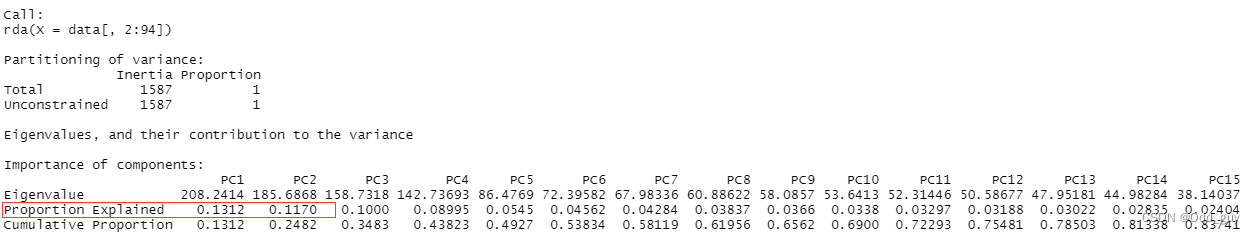
最后就是可视化:
# 生成坐标系
# 注意根据pca结果更改坐标轴名称
fig<-ordiplot(data_pca,type="none",xlab="PC1(13.12%)",ylab="PC2(11.70%)")# 将各样本点映射到坐标系
points(fig,"sites",pch=16,col="#FF4900FF",cex=1,select=data$site=="site_1")
points(fig,"sites",pch=16,col="#FF80FFFF",cex=1,select=data$site=="site_2")
points(fig,"sites",pch=16,col="#2A7AB7",cex=1,select=data$site=="site_3")
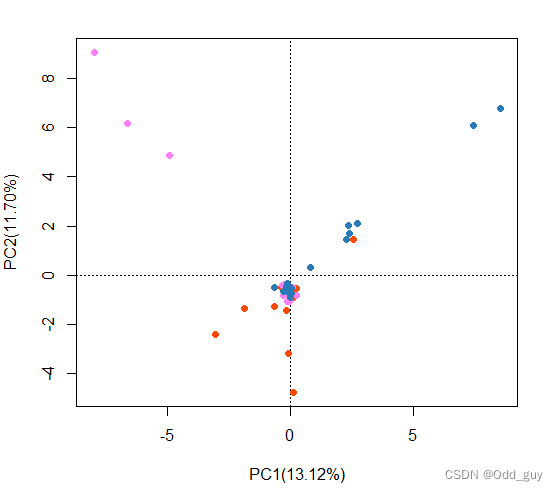
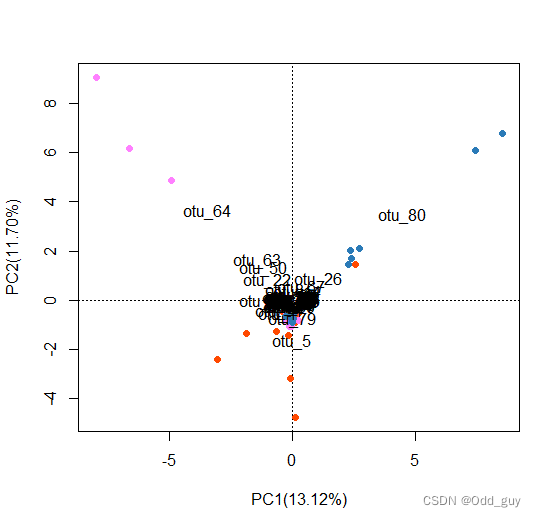
显示各OTUs信息:
# 显示各指标(列向量)
text(fig, "species", col="grey", cex=1)
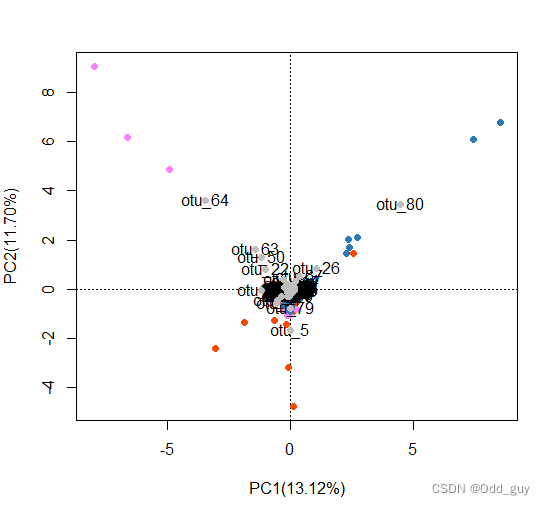
# 也可以显示列向量为点
points(fig, pch=16,"species", col="grey", cex=1)
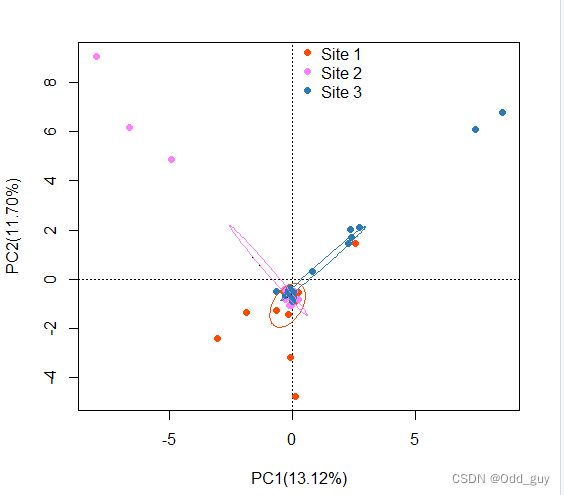
添加图例以及置信椭圆:
# 添加图例
legend(0,10, c("Site 1","Site 2", "Site 3"), text.col = c("black","black","black"),pch = c(16,16,16),col=c("#FF4900FF","#FF80FFFF","#2A7AB7"),cex=1,merge = F,bty = "n")# 读取分组信息
env<-read.csv("site.csv",header=T)# 添加置信椭圆
with(env,ordiellipse(data_pca,site,kind="se",col=c("#FF4900FF","#FF80FFFF","#2A7AB7"),conf = 0.99))
2 ggplot2
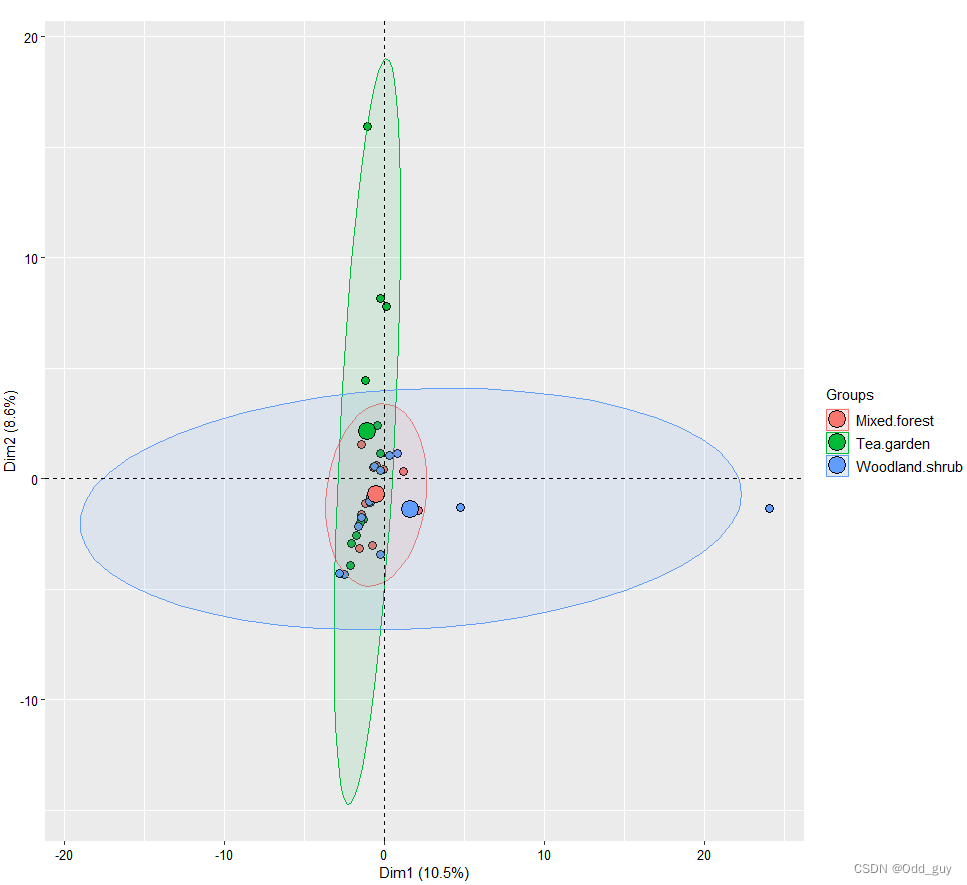
此次使用的数据是来自于三种生境的授粉昆虫多样性调查,每种生境分别做了12次重复。数据分析涉及的包为:ggplot2、factoextra、FactoMineR。相关的分析方法和原理与vegan类似:
library(ggplot2)
library(factoextra)
library(FactoMineR)# 1.
# 读取数据
setwd("D:/dir/CSDN/PCA/")
data<- read.csv("example2.csv")# 2.
# 降维,注意这提供了另一种切片方法
data_pca<- PCA(data[,-1], graph = FALSE)
summary(data_pca)# 3.
# 可视化
fviz_pca_ind(data_pca,geom.ind = "point", pointsize =3,pointshape = 21,fill.ind = data$site, addEllipses = TRUE, legend.title = "Groups",title="")+theme_grey() +theme(text=element_text(size=12,face="plain",color="black"),axis.title=element_text(size=11,face="plain",color="black"),axis.text = element_text(size=10,face="plain",color="black"),legend.title = element_text(size=11,face="plain",color="black"),legend.text = element_text(size=11,face="plain",color="black"),legend.background = element_blank(),legend.position="right")
#出图
#脚本参考自EasyCharts团队
最后,ggplot2做为强大的可视化工具,也可接受vegan包结果的投影,并使用ggplot的可视化思路进行绘图(之前做过,存脚本的磁盘坏了……)。