简介
MetaPhlAn2是分析微生物群落(细菌、古菌、真核生物和病毒)组成的工具,它在宏基因组研究中非常有用,只需一条完命令即可获得微生物的物种丰度信息(扩增子物种组成需要质控、拼接、拆样本、切除引物、比对等步骤,此软件居然分析宏基因组这么方便)。同时配合自带的脚本可进一步统计和可视化。
主页:http://segatalab.cibio.unitn.it/tools/metaphlan2/
MetaPhlAn2可以基于宏基因组数据,获得微生物群体中种水平精度的组成,包括细菌、古菌、真核生物和病毒。如果有株水平基因组的物种,也可以追踪和研究。
MetaPhlAn2整理了超过17000个参考基因组,包括13500个细菌和古菌,3500个病毒和110种真核生物,汇编整理了100万+类群特异的标记基因,可以实现:
精确的分类群分配
准确估计物种的相对丰度
种水平精度
株鉴定与追踪
超快的分析速度
软件 2015年发表于Nature Method,截止18年4月21日Google Scholar统计引用183次。该软件通讯作者为意大利特兰托大学的助理教授Nicola Segata的计算宏基因组实验室,己发表68篇文章,发表和维护的软件有14个,其中著名的有MetaPhlAn2, GraPhlAn, LEfSe, ShortBRED, MicroPITA, HUMAnN等。如果你对他不熟悉,那一定听説过他的博后老板Curtis Huttenhower(Picrust, Lefse通讯作者)。
实验室主页: http://segatalab.cibio.unitn.it
看看软件主要能干什么
分析人类微生物组数据,微生物和样本进行层级聚类
人类微生物组数据多时间点普氏菌属株水平的指纹
软件安装
优秀的是软件是一直在更新维护的,几年都没更新的软件,如果还有更好的选择就不建议使用。
此软件一直在更新维护,最近一次更新是两周前。
软件仓库:https://bitbucket.org/biobakery/metaphlan2/overview
依赖关系说明
软件依赖python2.7+和numpy包,同时居然支持python3。
如果使用Bowtie2结果为输入文件,则无任何依赖关系。
utils/metaphlan_hclust_heatmap.py脚本绘制热图时,依赖matplotlib包
如果想输出biom格式结果,需要安装R包biom
结果的进一步可视化,如使用hclust2和GraPhlAn请参考相关软件安装说明
下载和安装代码
请学习时下载安装最新版,如下代码仅为本文章作者学习此软件时的版本,仅供参考。主要包括下载、解压、添加环境变量。
wget https://bitbucket.org/biobakery/metaphlan2/get/default.zip
unzip default.zip
mv biobakery-metaphlan2-5175d8783801 metaphlan2
cd metaphlan2/
export PATH=`pwd`:`pwd`/utils:$PATH
export mpa_dir=`pwd`
metaphlan2.py -v # MetaPhlAn version 2.7.6 (5 March 2018)使用实例
定量物种谱
大家都知道扩增子分析前期处理步骤复杂,宏基因组分析应该更复杂,但此软件却是例外,操作简单,一条例命令从原始数据拿到物种谱(相当于标准化后的OTU表)。是不是很牛B。
而且作者考虑了不同用户的需求,有多种使用情况下都可用,下面多种方法根据自己的输入文件格式任选其一。
如果输入文件是fastq,需要依赖bowtie2安装好
metaphlan2.py metagenome.fastq --input_type fastq > profiled_metagenome.txt推荐输出bowtie2的比对结果,方便下次快速重新计算,并使用多线程-nproc
metaphlan2.py metagenome.fastq --bowtie2out metagenome.bowtie2.bz2 --nproc 9 --input_type fastq > profiled_metagenome.txt也可以使用bowtie2输出文件
metaphlan2.py metagenome.bowtie2.bz2 --nproc 5 --input_type bowtie2out > profiled_metagenome.txt也可以分别进行比对和定量
bowtie2 --sam-no-hd --sam-no-sq --no-unal --very-sensitive -S metagenome.sam -x ${mpa_dir}/databases/mpa_v20_m200 -U metagenome.fastq
metaphlan2.py metagenome.sam --input_type sam > profiled_metagenome.txt最常用的是使用双端压缩fastq文件,但并不考虑配对信息
metaphlan2.py --input_type fastq <(zcat metagenome_1.fq.gz metagenome_2.fq.gz) > profiled_metagenome.txt最好调用多线程,并输出比较结果
metaphlan2.py --bowtie2out metagenome.bowtie2.bz2 --nproc 9 --input_type fastq <(zcat metagenome_1.fq.gz metagenome_2.fq.gz) > profiled_metagenome1.txt第一次使用,程序会自己下载数据库至安装目录中,并进行校验、解压、解压、bowtie2建索引,根据网速和服务器性能可能需要很长时间1-N小时。
输出结果为各层级物种相对丰度值,有点像lefse的输入文件格式(方便lefse下游差异分析)
#SampleID Metaphlan2_Analysis
k__Bacteria 100.0
k__Bacteria|p__Actinobacteria 49.91104
k__Bacteria|p__Proteobacteria 46.00995
k__Bacteria|p__Firmicutes 2.45456
k__Bacteria|p__Bacteroidetes 0.99062
k__Bacteria|p__Cyanobacteria 0.63383
k__Bacteria|p__Actinobacteria|c__Actinobacteria 49.91104
k__Bacteria|p__Proteobacteria|c__Alphaproteobacteria 22.82115
k__Bacteria|p__Proteobacteria|c__Betaproteobacteria 16.60788多个样品批处理
假如我们有25-28一共4个样本,调用for循环并转后台并行处理
cd ~/project
for i in `seq 25 28`;do
metaphlan2.py --nproc 9 --input_type fastq <(zcat Sample_${i}.rmhost.1.fq.gz Sample_${i}.rmhost.2.fq.gz) > metaphlan2_${i}.txt &
done个人使用上面命令处理输入文件为压缩格式fastq时,偶尔会出错,帮修改为下面两步法分别比对和生成表格,更保险
for i in `seq 25 28`;do
bowtie2 -p 9 --sam-no-hd --sam-no-sq --no-unal --very-sensitive -S temp_${i}.sam -x ~/software/metaphlan2/databases/mpa_v20_m200 -U Sample_${i}.rmhost.1.fq.gz,Sample_${i}.rmhost.2.fq.gz &
donefor i in `seq 25 28`;do
metaphlan2.py temp_${i}.sam --input_type sam > metaphlan2_${i}.txt &
done样品物种谱合并
merge_metaphlan_tables.py命令可以将每个样品结果表合并,程序位于程序的utils目录中,请自行添加环境变量或使用绝对路径。合并时支持输入文件多个文件空格分隔,或使用通配符(如下)。可结合sed删除样本名中共有部分,精简样品名方便可视化简洁美观
merge_metaphlan_tables.py metaphlan2*.txt | sed 's/metaphlan2_//g' > merged_metaphlan2.txt获得了矩阵表,下面可以进行各种统计分析与可视化啦!
ID 25 26 27 28
#SampleID Metaphlan2_Analysis Metaphlan2_Analysis Metaphlan2_Analysis Metaphlan2_Analysis
k__Bacteria 100.0 100.0 100.0 100.0
k__Bacteria|p__Actinobacteria 49.91104 45.2479 54.37222 48.77918
k__Bacteria|p__Actinobacteria|c__Actinobacteria 49.91104 45.2479 54.37222 48.77918
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales 49.53809 44.96297 54.09146 48.77918
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Cellulomonadaceae 0.08247 0.064 0.0 0.0
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Cellulomonadaceae|g__Cellulomonas 0.08247 0.064 0.0 0.0
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Cellulomonadaceae|g__Cellulomonas|s__Cellulomonas_unclassified 0.08247 0.064 0.0 0.0绘制热图
metaphlan_hclust_heatmap.py -c bbcry --top 25 --minv 0.1 -s log --in merged_metaphlan2.txt --out abundance_heatmap.pdf
metaphlan_hclust_heatmap.py -c bbcry --top 25 --minv 0.1 -s log --in merged_metaphlan2.txt --out abundance_heatmap.pngc设置颜色方案,top设置物种数量,minv最大相对丰度,s标准化方法,文件名结尾可先pdf/png/svg三种图片格式。更多说明详见 metaphlan_hclust_heatmap.py -h
按各样本丰度绘制热图,显示前25种高丰度菌种类,样品默认采用bray_curtis距离聚类,微生物采用相关性距离聚类;数值采用对数10进行变换
自定义数据库
基于bowtie2数据库重建标记序列
bowtie2-inspect ~/software/metaphlan2/databases/mpa_v20_m200 > markers.fa追求你获得的maker至数据库
cat new_marker.fa >> markers.fa
bowtie2-build markers.fa ~/software/metaphlan2/databases/mpa_v21_m200并在python命令行中添加相应的物种注释信息,详见 https://bitbucket.org/biobakery/metaphlan2/overview#markdown-header-customizing-the-database
株水平群体基因组
此步配合StrainPhlAn软件,分析宏基因样本中菌株水平序列变异,输入序列样本,输出MSA多序列比对文件。StrainPhlAn采用RAxML构建进化树。
可以生成如下结果:
以HMP6个样本为例,分析粪拟杆菌Bacteroides caccae序列在株水平的变异
有需要有朋友可按如下教程安装相关软件并分析 https://bitbucket.org/biobakery/metaphlan2/overview#markdown-header-metagenomic-strain-level-population-genomics
GraPhlAn图
需要安装好GraPhlan
# 基于丰度表生成graphlan文件
export2graphlan.py --skip_rows 1,2 -i merged_metaphlan2.txt --tree merged_abundance.tree.txt --annotation merged_abundance.annot.txt --most_abundant 100 --abundance_threshold 1 --least_biomarkers 10 --annotations 5,6 --external_annotations 7 --min_clade_size 1
# 绘图
graphlan_annotate.py --annot merged_abundance.annot.txt merged_abundance.tree.txt merged_abundance.xml
graphlan.py --dpi 300 merged_abundance.xml merged_abundance.png --external_legends建议输出文件输出pdf,方便后期修改文字大小和内容
采用Graphlan展示物种组成
进一步学习
可参考metaphlan2官方教程
https://bitbucket.org/biobakery/biobakery/wiki/metaphlan2
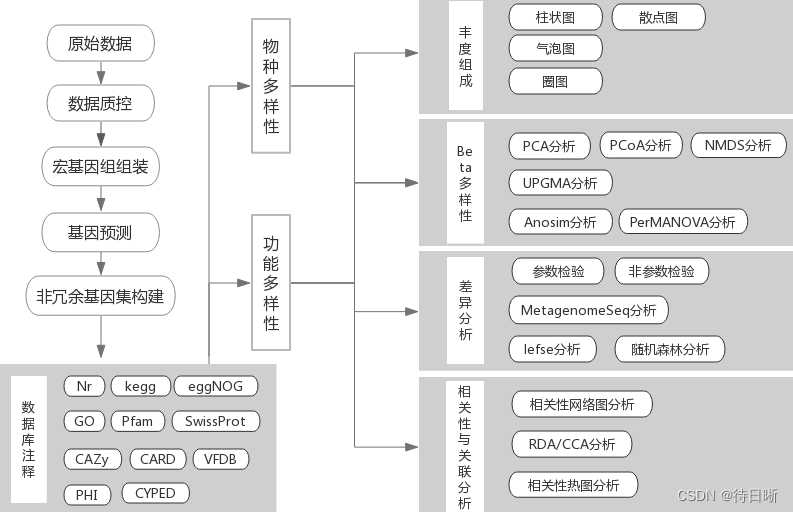
分析流程
参考文献
Duy Tin Truong, Eric Franzosa, Timothy L Tickle, Matthias Scholz, George Weingart, Edoardo Pasolli, Adrian Tett, Curtis Huttenhower, and Nicola Segata
MetaPhlAn2 for enhanced metagenomic taxonomic profiling
Nature Methods 12, 902–903 (2015)
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。
学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
点击阅读原文,跳转最新文章目录阅读