群体遗传学习笔记-测序技术学习
重测序技术简介
全基因组重测序(Resequencing)是对已知参考基因组序列的物种进行不同个体间的基因组测序,并在此基础上对个体或群体进行差异性分析。通过全基因组重测序,将不同梯度插入片段(Insert-Size)的测序文库结合短序列(Short-Reads)、双末端(Paired-End),可以找到大量的单核苷酸多态性位点(SNP)、拷贝数变异(Copy Number Variation,CNV)、插入缺失(InDel,Insertion/Deletion)、结构变异(Structure Variation,SV) 等变异信息,应用范围涉及临床医药研究、群体遗传学研究、关联分析、进化分析等众多领域。
原理
将特定组织或者细胞中的DNA进行随机打碎,构建片段为350bp或者500bp的文库,通过Illumina Hiseq对文库进行高通量测序,从而获得某一个个体所有DNA序列的信息。
全基因组数据分析的必要条件
- 所测物种的序列是有参考基因组的
- 所测序个体与参考基因组之间遗传差异性不大 (read 比对不上,很难找到SNP 等突变信息)
评价测序量的指标
测序深度是评价测序量的最重要指标。测序深度(Sequencing Depth):测序得到的碱基总量(bp)与基因组大小(Genome)的比值。
测序覆盖比例(Sequencing Coverage),指的是基因组上至少被检测到1次的区域,占整个基因组的比例。
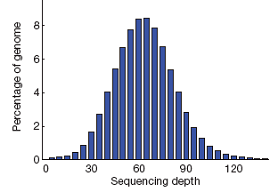
例如,在某1个样本测序的项目中,基因组平均测序深度为60X。基因组大部分区域的测序深度在60X左右,但同时依然有一小部分区域的测序深度低于3X(极低覆盖或没有覆盖)。
当然,这是理想条件下。在实际情况下的覆盖度,会低于理想值。主要是由于GC含量偏好,基因组完整性,个体差异,重复序列影响等。
此处输入图片的描述
如下图,在某1个样本测序的项目中,基因组平均测序深度为60X。基因组大部分区域的测序深度在60X左右,但同时依然有一小部分区域的测序深度低于3X(极低覆盖或没有覆盖)。
当然,这是理想条件下。在实际情况下的覆盖度,会低于理想值。主要是由于GC含量偏好,基因组完整性,个体差异,重复序列影响等。
测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降。重测序的个体,如果采用的是Paired-End,当测序深度达到10x时基因组的覆盖度已接近饱和,基因组覆盖度和测序错误率控制均得以保证。
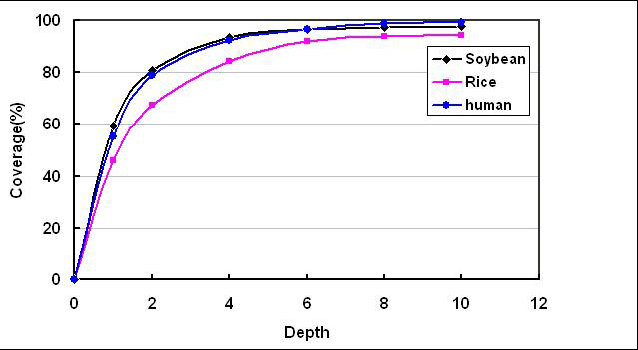
此处输入图片的描述
但SNP的检测率却没有达到饱和。这是由于当深度达到10X的时候,虽然基因组大部分区域已被覆盖,但在覆盖到的区域中,依然有相当多的区域深度小于34X。SNP检测的最低深度标准通常为34X。如果没有达到这个水准,则判断其不可靠,而在分析结果中不予接受。为了进一步减少低测序深度区域的比例,则需要进一步提高测序深度。只有测序深度达到30X的时候,SNP检测才会达到饱和
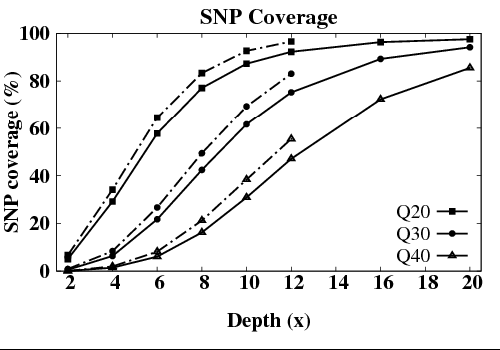
此处输入图片的描述
因此,可以根据我们的研究目的来选择相应的测序深度。
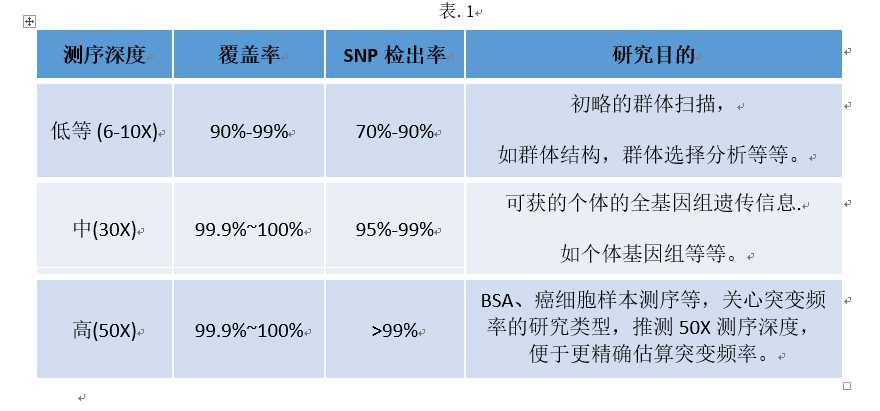
此处输入图片的描述
重测序应用
目前重测序技术已广泛应用于农学、医学等各个研究领域,包括性状相关候选基因筛选、动植物育种、单基因病筛查、癌症筛查等,快速准确,对育种和临床诊断有很好的指导作用。下面举例一些重测序常用的应用范围和主要思路。
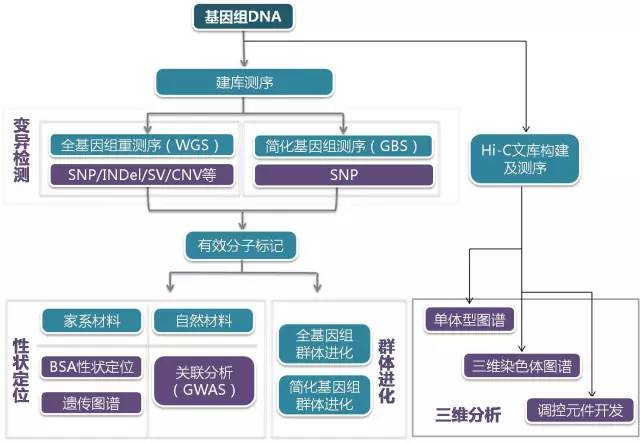
此处输入图片的描述
- 个体重测序,突变体检查 (对每个个体的位点进行扫描)
- 混池重测序,群体进化分析 (通过SNP进行后续分析)
- BSA,遗传图谱构建
- Hic-主要是做人类比较有用,这里不多说了。
动植物重测序文章思路
- GWAS与群体进化相结合
随着GWAS统计方法的不断完善,GWAS能够适用于大部分物种,将GWAS与群体进化结合分析为性状关键基因定位提供了一个新的思路。
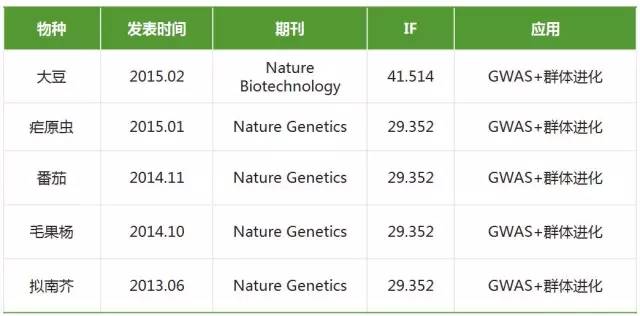
此处输入图片的描述
- GWAS与QTL定位相结合
连锁分析和关联分析在数量性状研究上都具有重要作用,它们在QTL定位的精度和广度、提供的信息量、统计分析方法等方面具有明显的互补性。
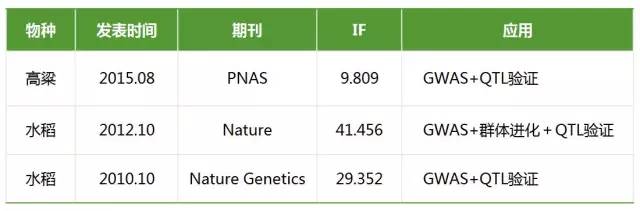
此处输入图片的描述
简化基因组测序
简单来说是对与限制性核酸内切酶识别位点相关的DNA进行高通量测序。
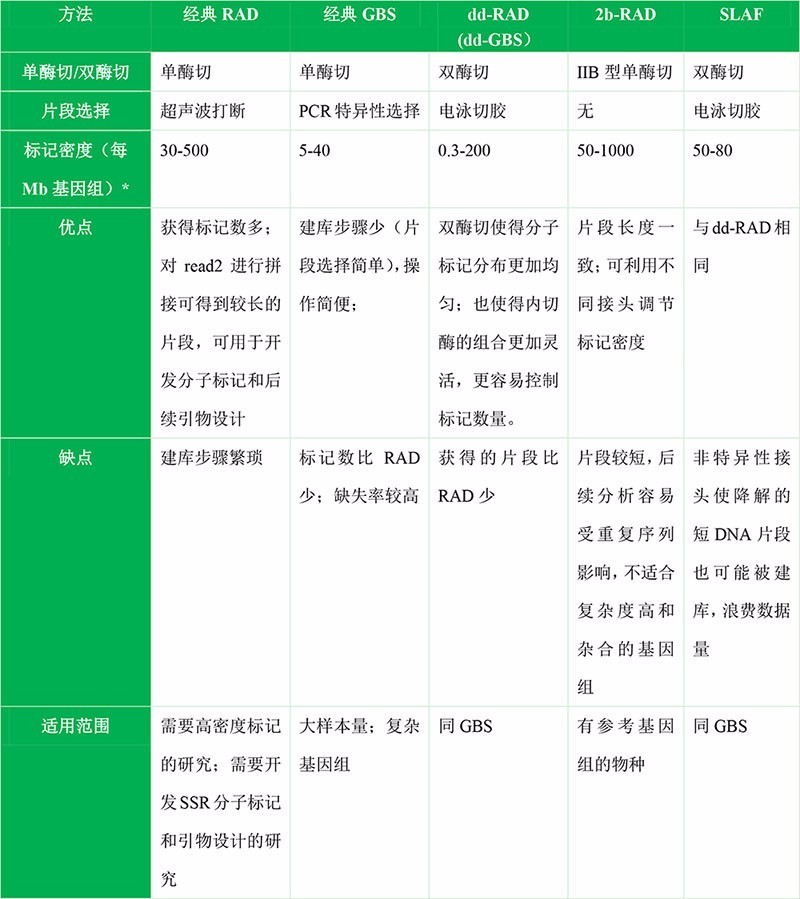
其实那么多种简化基因组方法的区别就在于单酶切还是双酶切、是否有随机打断、使用不同的内切酶、是否加barcode、接头设计等这些细节,本质还是一样的,就是对基因组进行酶切并对酶切片段进行测序。在这些方法中,RAD和GBS是使用最广泛的两种方法,2b-RAD,dd-RAD,SLAF等都是在这些方法的基础上在不同细节处改良。
RAD(Restriction site Associated DNA):是与限制性核酸内切酶识别位点相关的DNA。RAD方法对基因组DNA进行单酶切,然后对酶切片段超声波随机打断,因此测序得到的read1是位置对齐的,而read2是参差不齐的,因此可用于denovo聚类拼接,获得较长的contig,有利于开发SSR等分子标记。
GBS(Genotyping-By-Sequencing):是指通过测序进行基因分型。2011年由Elshire, R. J.提出2。GBS方法对基因组DNA进行单酶切,不需要超声波随机打断,而是利用PCR进行片段大小选择;并且对不同的样品加上不同的barcode,可对多达96个样品进行pooling建库,简化了建库步骤,因此比RAD成本更低。现在的GBS经过改良,普遍使用双酶切了,双酶切能够得到在基因组上分布更均一的酶切片段。双酶切的GBS,有时候又被称为dd-RAD。
dd-RAD(double-digest RAD,也可以称为dd-GBS):是双酶切的RAD,并且通过切胶来进行片段选择,2012年由Brant K.提出3。其实dd-RAD已经放弃了经典RAD的超声波片段化的策略,dd-RAD的建库流程和经典的GBS更为相似,所以下文我们也将之称为dd-GBS。 dd-RAD最大的优势在于,由于使用了两种内切酶处理,最终获得片段在基因组上的分布更加均一,从而提高了数据的有效性。由于目前的GBS方法普遍使用双酶切,并且用电泳切胶来取代PCR扩增来选择片段大小,因此dd-RAD几乎等同于目前的GBS方法了
不同方法之间的比较:
简化基因组的应用
简化基因组技术由于降低了基因组的复杂度、比全基因组重测序成本低,因此广泛应用于遗传图谱构建与QTL定位、群体进化分析、群体遗传分析、全基因组关联分析等研究领域。那么,这么多种方法,应该如何选择呢?概括来说可以从以下几方面考虑:
1. 所需标记数
不同的研究目的,所需的标记数量并不完全一样。通常,需要在全基因组范围内进行功能区间扫描和功能基因挖掘的研究,如全基因组关联分析和选择压力分析,就需要上万个高密度的分子标记,而系统发育关系、地理群体结构、基因流、系谱检测、连锁分析等研究的分子标记密度则不需要那么高,一般只需要几百到几千个分子标记足以完成分析。
对于基因定位的研究,不同的研究材料、作图群体,也会影响到需要的标记数目。例如利用自然群体进行全基因组关联分析,所需的标记数与物种的LD衰减距离相关,物种LD衰减得越快,所需的标记数就越多。又例如利用作图群体进行连锁作图QTL定位,所需的标记数与作图群体类型和群体大小有关。群体经历的世代越多(如RIL群体),群体越大,则重组事件越多,理论上提高标记密度可以有效提高遗传图谱的质量,所以所需的标记数越多。
因此,可先评估研究所需的标记数,再选择适合的简化基因组技术。RAD技术因为对所有酶切位点都检测,因此标记数要比GBS多,适合于需要标记密度高的研究,如选择压力分析。dd-GBS类的技术虽然收集的片段偏少,但标记分布更加均一,所以数据有效性更高;并且建库成本比RAD低,更适合大样品量的研究。
2.有无参考基因组
如果所研究物种没有参考基因组,那么RAD技术更合适,因为RAD技术可以利用不对齐的read2进行denovo拼接,再与read1拼接,可以得到长达400~500bp的片段,有利于SSR分子标记开发以及后续的引物设计。而2b-RAD由于片段过短,容易受重复序列干扰,且后期不利于设计引物验证测序得到的SNP。因此,2b-RAD不建议用在没有参考基因组的物种,和大的复杂的基因组上。
3.研究经费
简化基因组测序与全基因组重测序相比,由于只对酶切片段进行测序,因此在测序费用上大大下降。而由于目前的各种简化基因组技术都会使用barcode对多个样品进行混合建库,因此各方法间的建库成本差异已经不大。但RAD文库构建过程中有超声波打断步骤、dd-RAD需要使用Pippin Prep等仪器,因此成本还是要比GBS等高。在实际情况中,可根据具体样品数和研究经费选择合适的技术方法。
总的来说,RAD和GBS技术是使用最广泛的两种简化基因组技术,其他的技术方法都是在这两者的基础上的改进或细化。
基因分型芯片
基因分型芯片:利用已知的SNP位点侧翼的序列设计探针。探针固定在芯片上后,待测定样本的DNA与芯片杂交并扫描杂交荧光信号,从而鉴定这些探针位点(SNP位点)的基因型。最有代表性的品牌是illumina和affymetrix。
与简化组测序技术相比较
从以上比较,我们可以认为两种技术都是高性价比的大规模基因分型的方法。但最大的不同的是:
- 芯片基因分型本质上是对已知SNP多态位点的扫描,来确定样本在这个位点的基因型。(其实很多做人类医疗测序的都用的是芯片,人类的SNPs多态性信息已经比较完善了)那么,我们需要预先知道这个物种的基因组SNPs多态性信息(一般来源大规模重测序),然后筛选SNP设计芯片,才能进行后续的基因分型。打个比方:就是已经知道这个位置有个“坑”了,只是看看坑里到底是沙子还是水。所以芯片只能“分型”,不能“发现”。
- 简化基因组测序本质上还是测序,所以哪怕这个物种没有任何已知的SNPs信息,也能使用简化基因组测序进行检测。测序兼顾了“发现”和“分型”的功能。
两个技术的适用范围
1. 没有标准化芯片的非模式物种
遇到非主流的无标准化芯片的物种,测序无疑是最佳选择。兼顾了SNP的发现和基因型分型两个功能。
2. 有标准化芯片的模式物种
如果你研究的物种是人、猪、牛等这些物种,那么芯片公司的提供的芯片还是不错的选择的。毕竟这些芯片位点都是优化过的,基本是比较均匀地覆盖了相应物种的整个基因组。芯片的数据相对简单,后期数据的基本处理更简单。而测序数据,由于数据量大,后期数据的预处理复杂且需要较多计算资源。
那么,模式生物中是否抱定标准化芯片呢?也未必。主要还是两点:
1)芯片密度是否满足你的需求?
一些成熟的模式种,例如人,芯片密度都已经达到了兆级别。但对于某些农业种,芯片密度则还停留在较低的水平。例如:illumina玉米和绵羊的芯片,都停留在50k的密度水平,很久没有优化了。不过也可以理解他们的逻辑,反正芯片和测序仪都是他们家生产的。芯片密度不够?测序啊。哪怕是使用只对基因组一部分进行测序的简化基因组测序,也可以轻松获得几百k数量级的SNP标记。所以,对于芯片密度不够的情况下,测序是芯片很好的替代品。
2)对一些稀有位点的检测
由于设计芯片只使用群体中具有普遍性的多态位点,即这些位点都是在群体中高频出现的多态性位点。所以,对一般人群/种群进行普遍性的筛查的时候,没有太大的问题。但如果,我们研究的群体十分特殊(如,研究材料是比较偏的亚种),或研究目标就是低频甚至罕见位点的时候(癌症、家族遗传病),芯片就无能为力了——因为芯片上就没有这些位点的探针啊。
例如,你研究的是藏猪,那么猪的porcine 60k芯片效果就不会太好。因为芯片上的60k位点都是从常见的品种中筛查得到的,这些位点在藏猪这样的特殊亚种中可能多态性较差。而藏猪群体中普遍的多态性位点,标准化芯片上却没有。那么,这个时候简化基因组测序的效果会优于芯片。测序嘛,我测的是序列,管你SNPs稀有不稀有通通一网打尽。
总之,基因分型芯片和简化基因组测序,各有优缺点。在具体项目中,应该根据具体情况做选择。随着测序价格不断降低,测序的确会不断侵蚀芯片的市场空间。但芯片依然有其稳定、易标准化、效率高、成本容易控制等优点,在某些需要标准化的领域(例如:医疗诊断领域)有巨大的应用空间。